Rebetol 200mg Ribavirin Käyttö, sivuvaikutukset ja annostus. Hinta verkkoapteekissa. Geneeriset lääkkeet ilman reseptiä.
Mitä Rebetol 200mg on ja miten sitä käytetään?
Rebetol on reseptilääke, jota käytetään kroonisen hepatiitti C:n oireiden hoitoon. Rebetol 200 mg voidaan käyttää yksinään tai muiden lääkkeiden kanssa.
Rebetol kuuluu hepatiitti B/hepatiitti C -lääkkeiden luokkaan. RSV-agentit.
Ei tiedetä, onko Rebetol 200 mg turvallinen ja tehokas alle 3-vuotiaille lapsille.
Mitkä ovat Rebetolin mahdolliset sivuvaikutukset?
Rebetol voi aiheuttaa vakavia sivuvaikutuksia, mukaan lukien:
- nokkosihottuma,
- vaikeuksia hengittää,
- kasvojen, huulten, kielen tai kurkun turvotus,
- näköongelmia
- voimakas kipu ylävatsassa, joka leviää selkään,
- pahoinvointi,
- oksentelua,
- ripuli,
- uusi tai paheneva yskä,
- kuume,
- pistävä rintakipu,
- hengityksen vinkuminen,
- hengenahdistus,
- vakava masennus,
- ajatuksia itsensä vahingoittamisesta,
- ajatukset toisten satuttelusta,
- vaalea tai kellastunut iho,
- tumma virtsa,
- hämmennystä,
- heikkous,
- vilunväristykset,
- flunssan kaltaiset oireet,
- turvonneet ikenet,
- suun haavaumat,
- ihohaavat,
- helppo mustelma,
- epätavallinen verenvuoto ja
- huimaus
- Hakeudu välittömästi lääkärin hoitoon, jos sinulla on jokin yllä mainituista oireista.
- Rebetolin yleisimpiä sivuvaikutuksia ovat:
- pahoinvointi,
- oksentelua,
- ruokahalun menetys,
- kuume,
- vilunväristykset,
- vapina,
- alhainen verisolujen määrä,
- anemia,
- heikkous,
- väsymys,
- päänsärky,
- lihaskipu,
- mielialan muutoksia,
- ahdistus ja
- ärtyneisyys
Kerro lääkärille, jos sinulla on haittavaikutuksia, jotka häiritsevät sinua tai jotka eivät häviä.
Nämä eivät ole kaikkia Rebetolin mahdollisia sivuvaikutuksia. Lisätietoja saat lääkäriltäsi tai apteekista.
Soita lääkärillesi saadaksesi lääketieteellisiä neuvoja sivuvaikutuksista. Voit ilmoittaa sivuvaikutuksista FDA:lle numerossa 1-800-FDA-1088.
VAROITUS
VAKAVIEN HÄIRIÖIDEN JA RIBAVIRIINIIN LIITTYVIEN VAIKUTUSTEN RISKI
- REBETOL-monoterapia ei ole tehokas kroonisen hepatiitti C -virusinfektion hoidossa, eikä sitä tule käyttää yksinään tähän käyttöaiheeseen (ks. VAROITUKSET JA VAROTOIMET ).
- Ribaviriinin ensisijainen toksisuus on hemolyyttinen anemia. REBETOL 200 mg -hoitoon liittyvä anemia voi johtaa sydänsairauden pahenemiseen, mikä on johtanut kuolemaan johtaviin ja ei-fataalisiin sydäninfarkteihin. Potilaita, joilla on ollut merkittävä tai epästabiili sydänsairaus, ei tule hoitaa REBETOLilla [katso ANNOSTUS JA ANNOSTUS, VAROITUKSET JA VAROTOIMET sekä HAITTAVAIKUTUKSET].
- Merkittäviä teratogeenisia ja alkioita tappavia vaikutuksia on osoitettu kaikilla ribaviriinille altistuneilla eläinlajilla. Lisäksi ribaviriinin toistuvan annoksen puoliintumisaika on 12 päivää, joten se voi säilyä ei-plasmaosastoissa jopa 6 kuukautta. Siksi REBETOL-hoito on vasta-aiheinen raskaana oleville naisille ja raskaana olevien naisten miespuolisille puolisoille. Äärimmäistä varovaisuutta on noudatettava raskauden välttämiseksi hoidon aikana ja 6 kuukauden ajan hoidon päättymisen jälkeen sekä naispotilailla että REBETOL-hoitoa saavien miespotilaiden naispuolisilla kumppaneilla. Hoidon aikana ja 6 kuukauden hoidon jälkeisen seurantajakson aikana on käytettävä vähintään kahta luotettavaa tehokasta ehkäisyä [katso VASTA-AIHEET, VAROITUKSET JA VAROTOIMET, Käyttö tietyissä ryhmissä, Ei-kliininen toksikologia ja POTILASTIEDOT].
KUVAUS
REBETOL (ribaviriini) on synteettinen nukleosidianalogi (puriinianalogi). Ribaviriinin kemiallinen nimi on 1-β-D-ribofuranosyyli-1 H-1,2,4-triatsoli-3-karboksamidi ja sillä on seuraava rakennekaava (katso kuva 1).
Kuva 1: Rakennekaava
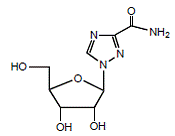
Ribaviriini on valkoinen, kiteinen jauhe. Se liukenee vapaasti veteen ja liukenee hieman vedettömään alkoholiin. Empiirinen kaava on C8H12N4O5 ja molekyylipaino on 244,21.
REBETOL-kapselit koostuvat valkoisesta jauheesta valkoisessa, läpinäkymättömässä gelatiinikapselissa. Jokainen kapseli sisältää 200 mg ribaviriinia ja inaktiivisia aineosia mikrokiteistä selluloosaa, laktoosimonohydraattia, kroskarmelloosinatriumia ja magnesiumstearaattia. Kapselin kuori koostuu gelatiinista, natriumlauryylisulfaatista, piidioksidista ja titaanidioksidista. Kapseli on painettu syötävällä sinisellä farmaseuttisella musteella, joka on valmistettu sellakasta, vedettömästä etyylialkoholista, isopropyylialkoholista, n-butyylialkoholista, propyleeniglykolista, ammoniumhydroksidista ja FD&C Blue #2 -alumiinilakasta.
REBETOL 200 mg oraaliliuos on kirkas, väritön tai vaaleankeltainen purukumimakuinen neste. Jokainen millilitra liuosta sisältää 40 mg ribaviriinia ja inaktiivisia aineosia sakkaroosia, glyseriiniä, sorbitolia, propyleeniglykolia, natriumsitraattia, sitruunahappoa, natriumbentsoaattia, luonnollista ja keinotekoista aromia purukumille #15864 ja vettä.
INDIKAATIOT
Krooninen hepatiitti C (CHC)
REBETOL® (ribaviriini) yhdessä interferoni alfa-2b:n (pegyloitu ja pegyloitumaton) kanssa on tarkoitettu kroonisen hepatiitti C:n (CHC) hoitoon 3-vuotiaille ja sitä vanhemmille potilaille, joilla on kompensoitunut maksasairaus [ks. VAROITUKSET JA VAROTOIMET , ja Käyttö tietyissä populaatioissa ].
Seuraavat seikat tulee ottaa huomioon aloitettaessa REBETOL-yhdistelmähoitoa PegIntron®- tai INTRON A®:n kanssa:
- Nämä käyttöaiheet perustuvat havaitsemattoman HCV-RNA:n saavuttamiseen 24 tai 48 viikon hoidon jälkeen ja jatkuvan virologisen vasteen (SVR) ylläpitämiseen 24 viikkoa viimeisen annoksen jälkeen.
- Yhdistelmähoitoa REBETOL/PegIntronilla suositellaan REBETOL/INTRON A:han verrattuna, koska tämä yhdistelmä tarjoaa huomattavasti paremman vasteen [katso Kliiniset tutkimukset ].
- Potilaat, joilla on seuraavat ominaisuudet, hyötyvät epätodennäköisemmin uusintahoidosta epäonnistuneen hoitojakson jälkeen: aikaisempi vaste, aiempi pegyloitu interferonihoito, merkittävä siltafibroosi tai kirroosi ja genotyypin 1 infektio [ks. Kliiniset tutkimukset ].
- Turvallisuutta ja tehoa koskevia tietoja yli vuoden kestäneestä hoidosta ei ole saatavilla.
ANNOSTELU JA HALLINNOINTI
REBETOL-kapseleita ei saa missään tapauksessa avata, murskata tai rikkoa. REBETOL 200mg tulee ottaa ruoan kanssa [katso KLIININEN FARMAKOLOGIA ]. REBETOLia ei tule käyttää potilaille, joiden kreatiniinipuhdistuma on alle 50 ml/min.
REBETOL/PegIntron-yhdistelmähoito
Aikuiset potilaat
Suositeltu PegIntron-annos on 1,5 mcg/kg/viikko ihonalaisesti yhdessä 800–1400 mg REBETOL 200 mg kapselin kanssa suun kautta potilaan painon perusteella (ks. taulukko 1). Pistettävän PegIntron-annoksen määrä riippuu PegIntronin vahvuudesta ja potilaan painosta. Katso lisätietoja annostelusta PegIntronin etiketistä.
Hoidon kesto – Alfa-interferonia aiemmin saaneet potilaat
Hoidon kesto potilailla, joilla on genotyyppi 1, on 48 viikkoa. Hoidon lopettamista tulee harkita potilailla, jotka eivät saavuta vähintään 2 log10:n pudotusta tai HCV-RNA:n häviämistä 12 viikon kohdalla tai jos HCV-RNA on edelleen havaittavissa 24 viikon hoidon jälkeen. Potilaita, joilla on genotyyppi 2 ja 3, tulee hoitaa 24 viikon ajan.
Hoidon kesto – PegIntron/REBETOL-hoito uudelleen, jos hoito epäonnistui
Hoidon kesto potilailla, joiden hoito aiemmin epäonnistui, on 48 viikkoa HCV-genotyypistä riippumatta. Uudelleen hoidetut potilaat, jotka eivät saavuta havaitsematonta HCV-RNA:ta hoitoviikolla 12 tai joiden HCV-RNA on edelleen havaittavissa 24 viikon hoidon jälkeen, on erittäin epätodennäköistä, että saavutetaan SVR, ja hoidon lopettamista tulee harkita [ks. Kliiniset tutkimukset ].
Pediatriset potilaat
Lapsipotilaiden annostus määräytyy PegIntronin kehon pinta-alan ja REBETOLin painon mukaan. Suositeltu PegIntron-annos on 60 mcg/m²/viikko ihon alle yhdistettynä 15 mg/kg/vrk REBETOL 200 mg suun kautta kahteen jaettuun annokseen (katso taulukko 2) 3–17-vuotiaille lapsipotilaille. Potilaiden, jotka täyttävät 18 vuotta, kun he saavat PegIntron/REBETOL 200 mg -valmistetta, tulee jatkaa lasten annostusohjelmaa. Hoidon kesto potilailla, joilla on genotyyppi 1, on 48 viikkoa. Potilaita, joilla on genotyyppi 2 ja 3, tulee hoitaa 24 viikon ajan.
REBETOL/INTRON A yhdistelmähoito
Aikuiset
Hoidon kesto – Alfa-interferonia aiemmin saaneet potilaat
INTRON A:n suositeltu annos on 3 miljoonaa IU kolme kertaa viikossa ihon alle. Suositeltu REBETOL-kapseleiden annos riippuu potilaan painosta (katso taulukko 3). Suositeltu hoidon kesto potilailla, joita ei ole aiemmin hoidettu interferonilla, on 24-48 viikkoa. Hoidon kesto tulee yksilöidä potilaalle riippuen sairauden perusominaisuuksista, hoitovasteesta ja hoito-ohjelman siedettävyydestä [ks. KÄYTTÖAIHEET JA KÄYTTÖ , HAITTAVAIKUTUKSET , ja Kliiniset tutkimukset ]. 24 viikon hoidon jälkeen virologinen vaste on arvioitava. Hoidon lopettamista tulee harkita jokaisella potilaalla, jonka HCV-RNA ei ole saavuttanut määrityksen havainnointirajan alapuolella olevaa arvoa 24 viikkoon mennessä. Yli 48 viikkoa kestäneen hoidon turvallisuudesta ja tehosta ei ole olemassa tietoja aiemmin hoitamattomilla potilailla.
Hoidon kesto – Uusintahoito INTRON A/REBETOLilla uusiutuneilla potilailla
Suositeltu hoidon kesto on 24 viikkoa potilailla, jotka uusiutuvat pegyloitumattoman interferonin monoterapian jälkeen.
Pediatria
Suositeltu REBETOL-annos on 15 mg/kg päivässä suun kautta (jaettu annos AM ja PM). Katso taulukko 2 lapsille tarkoitetun REBETOLin annostelusta yhdessä INTRON A:n kanssa. INTRON A injektioon 25–61 kg:n painoinen on 3 miljoonaa IU/m² kolmesti viikossa ihonalaisesti. Katso aikuisten annostustaulukosta yli 61 kg painoisille.
Suositeltu hoidon kesto on 48 viikkoa lapsipotilailla, joilla on genotyyppi 1. Virologinen vaste on arvioitava 24 viikon hoidon jälkeen. Hoidon lopettamista tulee harkita jokaisella potilaalla, jonka HCV-RNA ei ole tähän mennessä saavuttanut määrityksen havaitsemisrajan alapuolella. Suositeltu hoidon kesto lapsipotilailla, joilla on genotyyppi 2/3, on 24 viikkoa.
Laboratoriotestit
Seuraavia laboratoriotutkimuksia suositellaan kaikille REBETOL 200 mg:lla hoidetuille potilaille ennen hoidon aloittamista ja sen jälkeen määräajoin.
Normaalit hematologiset testit - mukaan lukien hemoglobiini (esihoito, hoidon viikko 2 ja viikko 4 ja kliinisesti tarkoituksenmukaisesti [ks. VAROITUKSET JA VAROTOIMET ], täydellinen ja differentiaalinen valkosolujen määrä ja verihiutaleiden määrä.
- Verikemiat - maksan toimintakokeet ja TSH.
- Raskaus - mukaan lukien kuukausittainen seuranta hedelmällisessä iässä oleville naisille.
- EKG [katso VAROITUKSET JA VAROTOIMET ].
Annoksen muutokset
Jos vakavia haittavaikutuksia tai laboratorioarvojen poikkeavuuksia kehittyy REBETOL/INTRON A-yhdistelmähoidon tai REBETOL/PegIntron-hoidon aikana, muuta annosta tai keskeytä annosta, kunnes haittavaikutus vähenee tai vaikeutuu [ks. VAROITUKSET JA VAROTOIMET ]. Jos intoleranssi jatkuu annoksen muuttamisen jälkeen, yhdistelmähoito on lopetettava. PegIntron-annosta pienennetään REBETOL/PegIntron-yhdistelmähoitoa saavilla aikuispotilailla kaksivaiheisessa prosessissa alkuperäisestä aloitusannoksesta 1,5 mikrogrammaa/kg/viikko 1 mikrogrammaan/kg/viikko ja sitten 0,5 mikrogrammaan/kg/viikko. , tarvittaessa. Katso PegIntronin pakkausmerkinnöistä lisätietoja PegIntron-annoksen pienentämisestä.
Aikuisten yhdistelmähoidon tutkimuksessa 2 annosta pienennettiin 42 %:lla potilaista, jotka saivat PegIntronia 1,5 mikrogrammaa/kg ja REBETOLia 800 mg päivässä, mukaan lukien 57 %:lla potilaista, jotka painoivat 60 kg tai vähemmän. Tutkimuksessa 4 16 %:lla potilaista PegIntron-annosta pienennettiin 1 mikrogrammaan/kg yhdessä REBETOLin kanssa, ja lisäksi 4 % vaati toisen PegIntron-annoksen pienentämisen arvoon 0,5 mikrogrammaa/kg haittatapahtumien vuoksi [ks. HAITTAVAIKUTUKSET ].
Lapsipotilaiden annosta pienennetään muuttamalla suositeltua PegIntron-annosta kaksivaiheisessa prosessissa alkuperäisestä aloitusannoksesta 60 mcg/m²/viikko 40 mikrogrammaan/m²/viikko ja sitten 20 mikrogrammaan/m²/viikko, jos tarvitaan (katso taulukko 4). Lasten yhdistelmähoitotutkimuksessa annosta pienennettiin 25 %:lla potilaista, jotka saivat PegIntronia 60 mcg/m² viikossa ja REBETOLia 15 mg/kg päivässä. Lapsipotilaiden annosta pienennetään muuttamalla suositeltua REBETOL-annosta alkuperäisestä aloitusannoksesta 15 mg/kg/vrk kaksivaiheisessa prosessissa 12 mg/kg/vrk, sitten 8 mg/kg/vrk, jos tarpeen. katso taulukko 4).
REBETOL 200 mg:aa ei tule käyttää potilaille, joiden kreatiniinipuhdistuma on alle 50 ml/min. Potilaita, joilla on munuaisten vajaatoiminta ja yli 50-vuotiaita, tulee seurata huolellisesti anemian kehittymisen suhteen [ks. VAROITUKSET JA VAROTOIMET , Käyttö tietyissä populaatioissa , ja KLIININEN FARMAKOLOGIA ].
REBETOL 200 mg:aa tulee antaa varoen potilaille, joilla on ennestään sydänsairaus. Potilaat tulee arvioida ennen hoidon aloittamista, ja heitä tulee seurata asianmukaisesti hoidon aikana. Jos kardiovaskulaarinen tila heikkenee, hoito on lopetettava [katso VAROITUKSET JA VAROTOIMET ].
Jos potilaalla on ollut vakaa sydän- ja verisuonisairaus, annosta on pienennettävä pysyvästi, jos hemoglobiini laskee vähintään 2 g/dl minkä tahansa 4 viikon aikana. Lisäksi, jos hemoglobiini on alle 12 g/dl 4 viikon jälkeen pienennetyllä annoksella, potilaan tulee lopettaa yhdistelmähoito näillä sydänpotilailla.
On suositeltavaa, että potilaalle, jonka hemoglobiinitaso laskee alle 10 g/dl, REBETOL-annosta muutetaan tai se lopetetaan taulukon 4 mukaisesti [ks. VAROITUKSET JA VAROTOIMET ].
Katso INTRON A- tai PegIntron-merkinnöistä lisätietoja INTRON A- tai PegIntron-annoksen pienentämisestä.
Annostelun lopettaminen
Aikuiset
HCV-genotyypin 1 potilailla, jotka eivät ole aiemmin saaneet interferonia alfaa ja saavat PegIntronia yhdistelmänä ribaviriinin kanssa, hoidon lopettamista suositellaan, jos HCV-RNA:n pudotus tai häviö ei ole vähintään 2 log10 12 viikon hoidon jälkeen tai jos HCV-RNA tasot pysyvät havaittavissa 24 viikon hoidon jälkeen. Genotyypistä riippumatta aiemmin hoidetuilla potilailla, joilla on havaittavissa oleva HCV-RNA viikolla 12 tai 24, on erittäin epätodennäköistä, että SVR saavutetaan, ja hoidon lopettamista tulee harkita.
Pediatria (3-17-vuotiaat)
On suositeltavaa, että potilaat, jotka saavat PegIntron/REBETOL-yhdistelmää (pois lukien HCV genotyypit 2 ja 3), lopetetaan 12 viikon kohdalla, jos heidän hoitoviikon 12 HCV-RNA putosi alle 2 log10 verrattuna esihoitoon tai 24 viikon kohdalla, jos heillä on havaittavissa olevia oireita. HCV-RNA hoidon aikana viikolla 24.
MITEN TOIMITETAAN
Annostusmuodot ja vahvuudet
REBETOL 200mg Kapselit 200mg
REBETOL 200mg oraaliliuos 40 mg/ml
Varastointi ja käsittely
REBETOL 200 mg kapselit ovat valkoisia, läpinäkymättömiä kapseleita, joissa on REBETOL, 200 mg, ja Schering Corporationin logo painettu kapselin kuoreen; kapselit on pakattu pulloon, joka sisältää 56 kapselia ( NDC 0085-1351-05), 70 kapselia ( NDC 0085-1385-07) ja 84 kapselia ( NDC 0085-1194-03).
REBETOL 200 mg oraaliliuos 40 mg per ml on kirkas, väritön tai vaaleankeltainen purukumimakuinen neste, ja se on pakattu 4 unssin ruskeisiin lasipulloihin (100 ml/pullo), joissa on lapsiturvallinen suljin ( NDC 0085-1318-01).
REBETOL-kapseleiden pullo tulee säilyttää 25 °C:ssa (77 °F); retket sallitaan 15-30°C:een (59-86°F) [katso USP-ohjattu huonelämpötila ].
REBETOL 200 mg oraaliliuosta tulee säilyttää 2-8°C:ssa (36-46°F) tai 25°C:ssa (77°F); retket sallitaan 15-30°C:een (59-86°F) [katso USP-ohjattu huonelämpötila ].
REBETOL 200 mg oraaliliuos valmistettu: Merck Sharp & Dohme Corp., Whitehouse Stationin tytäryhtiö, NJ 08889, USA. Valmistaja: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Kanada REBETOL-kapselit valmistaja: Merck Sharp & Dohme Corp., Whitehouse Stationin tytäryhtiö, NJ 08889, USA. Tarkistettu: joulukuuta 2014.
SIVUVAIKUTUKSET
Kliiniset tutkimukset REBETOLilla yhdessä PegIntronin tai INTRON A:n kanssa on suoritettu yli 7800:lla 3–76-vuotiaalla henkilöllä.
Ribaviriinin ensisijainen toksisuus on hemolyyttinen anemia. Hemoglobiinitasot laskivat ensimmäisten 1-2 viikon aikana oraalisen hoidon aikana. Anemiaan liittyviä sydän- ja keuhkoreaktioita esiintyi noin 10 %:lla potilaista [katso VAROITUKSET JA VAROTOIMET ].
Yli 96 % kaikista kliinisissä tutkimuksissa koehenkilöistä koki yhden tai useamman haittavaikutuksen. Yleisimmin raportoituja haittavaikutuksia aikuisilla, jotka saivat PegIntronia tai INTRON A:ta yhdessä REBETOL 200 mg:n kanssa, olivat pistoskohdan tulehdus/reaktio, väsymys/astenia, päänsärky, jäykkyys, kuume, pahoinvointi, lihaskipu ja ahdistuneisuus/emotionaalinen labilisuus/ärtyneisyys. Yleisimmät haittavaikutukset 3-vuotiailla ja sitä vanhemmilla lapsilla, jotka saivat REBETOL 200 mg yhdistelmänä PegIntronin tai INTRON A:n kanssa, olivat kuume, päänsärky, neutropenia, väsymys, anoreksia, pistoskohdan punoitus ja oksentelu.
Haittavaikutukset-osiossa viitataan seuraaviin kliinisiin tutkimuksiin:
- REBETOL/PegIntron-yhdistelmähoitotutkimukset:
- Kliininen tutkimus 1 – arvioitu PegIntron-monoterapia (ei kuvattu tarkemmin tässä etiketissä; katso lisätietoja tästä tutkimuksesta PegIntron-merkinnöistä).
- Tutkimus 2 – arvioitiin REBETOL 800 mg/vrk tasainen annos yhdessä 1,5 mikrog/kg/viikko PegIntronin tai INTRON A:n kanssa.
- Tutkimus 3 – arvioitiin PegIntron/painopohjainen REBETOL 200 mg yhdessä PegIntron/tasainen annos REBETOL 200 mg hoito-ohjelman kanssa.
- Tutkimus 4 – verrattiin kahta PegIntron-annosta (1,5 mcg/kg/viikko ja 1 mcg/kg/viikko) yhdessä REBETOLin kanssa ja kolmatta hoitoryhmää, joka sai Pegasys® (180 mcg/viikko) / Copegus® (1000-1200 mg/vrk) ).
- Tutkimus 5 – arvioitiin PegIntronia (1,5 mikrog/kg/viikko) yhdessä painoon perustuvan REBETOLin kanssa potilailla, joilla hoito epäonnistui.
- PegIntron/REBETOL 200 mg yhdistelmähoito lapsipotilailla
- REBETOL/INTRON A yhdistelmähoitokokeet aikuisille ja lapsille
Vakavia haittavaikutuksia on esiintynyt noin 12 %:lla potilaista kliinisissä tutkimuksissa PegIntronilla yhdessä REBETOLin kanssa tai ilman sitä [ks. LAATIKKO VAROITUS , VAROITUKSET JA VAROTOIMET ]. PegIntronilla ja REBETOL 200 mg:lla hoidetuilla potilailla yleisimmät vakavat tapahtumat olivat masennus ja itsemurha-ajatukset [ks. VAROITUKSET JA VAROTOIMET ], joista kukin esiintyy alle 1 %:n taajuudella. Itsemurha-ajatuksia tai -yrityksiä esiintyi useammin lapsipotilailla, pääasiassa nuorilla, verrattuna aikuisiin potilaisiin (2,4 % vs. 1 %) hoidon ja hoidon ulkopuolisen seurannan aikana [ks. VAROITUKSET JA VAROTOIMET ]. Yleisin kuolemaan johtava reaktio PegIntronilla ja REBETOL 200 mg:lla hoidetuilla henkilöillä oli sydämenpysähdys, itsemurha-ajatukset ja itsemurhayritys [ks. VAROITUKSET JA VAROTOIMET ], kaikkia esiintyy alle 1 %:lla koehenkilöistä.
Koska kliiniset tutkimukset suoritetaan hyvin vaihtelevissa olosuhteissa, lääkkeen kliinisissä tutkimuksissa havaittuja haittavaikutuksia ei voida verrata suoraan toisen lääkkeen kliinisissä tutkimuksissa havaittuihin määriin, eivätkä ne välttämättä kuvasta kliinisessä käytännössä havaittuja määriä.
Kokemus kliinisistä kokeista – REBETOL/PegIntron-yhdistelmähoito
Aikuiset aiheet
Haittavaikutukset, jotka ilmenivät kliinisessä tutkimuksessa yli 5 %:n ilmaantuvuudella, on ilmoitettu REBETOL/PegIntron-yhdistelmähoidon (tutkimus 2) hoitoryhmässä taulukossa 5.
Taulukossa 6 on yhteenveto hoitoon liittyvistä haittavaikutuksista tutkimuksessa 4, joita esiintyi vähintään 10 %:lla.
Vakavien haittavaikutusten ilmaantuvuus oli vertailukelpoinen kaikissa tutkimuksissa. Tutkimuksessa 3 ilmoitettiin samanlainen vakavien haittavaikutusten ilmaantuvuus painoon perustuvassa REBETOL-ryhmässä (12 %) ja tasa-annoksisessa REBETOL-ohjelmassa. Tutkimuksessa 2 vakavien haittavaikutusten ilmaantuvuus oli 17 % PegIntron/REBETOL-ryhmissä ja 14 % INTRON A/REBETOL -ryhmässä.
Monissa mutta ei kaikissa tapauksissa haittavaikutukset hävisivät annoksen pienentämisen tai hoidon lopettamisen jälkeen. Jotkut koehenkilöt kokivat jatkuvia tai uusia vakavia haittavaikutuksia 6 kuukauden seurantajakson aikana. Tutkimuksessa 2 monet koehenkilöt kokivat haittavaikutuksia useita kuukausia hoidon lopettamisen jälkeen. Kuuden kuukauden seurantajakson loppuun mennessä jatkuvien haittavaikutusten ilmaantuvuus keholuokittain PegIntron 1,5/REBETOL 200 mg -ryhmässä oli 33 % (psykiatriset), 20 % (tuki- ja liikuntaelimistöt) ja 10 % (umpieritys- ja GI:lle). Noin 10-15 %:lla potilaista painonpudotus, väsymys ja päänsärky eivät olleet hävinneet.
Näissä kliinisissä tutkimuksissa hoidon tai seurannan aikana on tapahtunut 31 koehenkilön kuolemaa. Tutkimuksessa 1 PegIntron-monoterapiaa saaneella henkilöllä tapahtui yksi itsemurha ja INTRON A -monoterapiaa saaneiden henkilöiden joukossa kaksi kuolemaa (1 murha/itsemurha ja 1 äkkikuolema). Tutkimuksessa 2 PegIntron/REBETOL 200 mg yhdistelmähoitoa saaneella henkilöllä oli yksi itsemurha; ja yksi henkilö kuoli INTRON A/REBETOL 200 mg -ryhmässä (moottoriajoneuvo-onnettomuus). Tutkimuksessa 3 oli 14 kuolemaa, joista 2 oli todennäköisiä itsemurhia ja 1 oli selittämätön kuolema henkilöllä, jolla oli vastaava lääketieteellinen masennus. Tutkimuksessa 4 tapahtui 12 kuolemaa, joista 6 sattui koehenkilöillä, jotka saivat PegIntron/REBETOL-yhdistelmähoitoa, 5 PegIntron 1,5 mikrog/REBETOL -haarassa (N = 1019) ja 1 PegIntron 1 mikrog/REBETOL -haarassa (N = 1016), ja joista 6 esiintyi Pegasys/Copegus-potilailla (N=1035); 3 itsemurhaa tapahtui hoidon ulkopuolisen seurantajakson aikana henkilöillä, jotka saivat PegIntron (1,5 mikrogrammaa/kg)/REBETOL 200 mg -yhdistelmähoitoa.
Tutkimuksissa 1 ja 2 10–14 % potilaista, jotka saivat PegIntronia yksinään tai yhdessä REBETOL 200 mg:n kanssa, keskeytti hoidon, kun taas 6 % sai INTRON A:ta yksinään ja 13 % INTRON A:ta yhdessä REBETOLin kanssa. Vastaavasti tutkimuksessa 3 15 % potilaista, jotka saivat PegIntronia yhdessä painoon perustuvan REBETOLin kanssa, ja 14 % potilaista, jotka saivat PegIntronia ja 200 mg:n vakioannos REBETOLia, keskeytti hoidon haittavaikutuksen vuoksi. Yleisimmät syyt hoidon keskeyttämiseen liittyivät psykiatristen, systeemisten (esim. väsymys, päänsärky) tai maha-suolikanavan haittavaikutusten tunnettuihin interferonivaikutuksiin. Tutkimuksessa 4 13 %:lla potilaista PegIntron 1,5 mikrog/REBETOL 200 mg -haarassa, 10 %:lla PegIntron 1 mikrog/REBETOL 200 mg -haarassa ja 13 %:lla Pegasys 180 mikrog/Copegus -haarassa haitallisten tapahtumien vuoksi.
Tutkimuksessa 2 haittavaikutuksista johtuvia annosta pienennettiin 42 %:lla potilaista, jotka saivat PegIntron (1,5 mikrogrammaa/kg)/REBETOL 200 mg -valmistetta, ja 34 %:lla potilaista, jotka saivat INTRON A/REBETOLia. Suurin osa PegIntronia (1,5 mikrog/kg)/REBETOLia saaneista potilaista (57 %), jotka painoivat 60 kg tai vähemmän, vaativat annoksen pienentämistä. Interferonin vähentäminen oli annoksesta riippuvaa (PegIntron 1,5 mcg/kg suurempi kuin PegIntron 0,5 mcg/kg tai INTRON A), vastaavasti 40 %, 27 % ja 28 %. REBETOL 200 mg:n annoksen pienennys oli samanlainen kaikissa kolmessa ryhmässä, 33-35 %. Yleisimmät syyt annoksen muuttamiseen olivat neutropenia (18 %) tai anemia (9 %) (ks Laboratorioarvot ). Muita yleisiä syitä olivat masennus, väsymys, pahoinvointi ja trombosytopenia. Tutkimuksessa 3 haittavaikutuksista johtuvia annosmuutoksia esiintyi useammin painoon perustuvalla annostelulla (WBD) verrattuna tasaiseen annostukseen (29 % ja 23 %). Tutkimuksessa 4 16 %:lla koehenkilöistä PegIntron-annosta pienennettiin 1 mikrogrammaan/kg yhdessä REBETOL 200 mg:n kanssa, ja lisäksi 4 % vaati toisen PegIntron-annoksen pienentämisen arvoon 0,5 mikrogrammaa/kg haittatapahtumien vuoksi verrattuna 15 %:iin. Pegasys/Copegus-haarassa, jotka tarvitsivat annoksen pienentämistä 135 mikrogrammaan/viikko Pegasys-ryhmässä, ja lisäksi 7 % Pegasys/Copegus-haarassa vaati toisen annoksen pienentämistä 90 mikrogrammaan/viikko Pegasysin kanssa.
PegIntron/REBETOL 200 mg yhdistelmätutkimuksissa yleisimmät haittavaikutukset olivat psykiatriset, joita esiintyi 77 %:lla tutkimuksen 2 koehenkilöistä ja 68-69 %:lla tutkimuksen 3 koehenkilöistä. Näitä psykiatrisia haittavaikutuksia olivat yleisimmin masennus, ärtyneisyys ja unettomuus, joista kutakin raportoi noin 30–40 % koehenkilöistä kaikissa hoitoryhmissä. Itsemurhakäyttäytymistä (ajatuksia, yrityksiä ja itsemurhia) esiintyi 2 %:lla kaikista koehenkilöistä hoidon aikana tai seurannan aikana hoidon lopettamisen jälkeen [ks. VAROITUKSET JA VAROTOIMET ]. Tutkimuksessa 4 psykiatrisia haittavaikutuksia esiintyi 58 %:lla PegIntron 1,5 mcg/REBETOL 200 mg -haarassa, 55 %:lla PegIntron 1 mcg/REBETOL 200 mg -haarassa ja 57 %:lla potilaista PegIntron 1,5 mcg/REBETOL 200 mg -haarassa ja 57 %:lla koehenkilöistä PegIntron 1,5 mcg/REBETOL 200 mg ryhmässä. .
PegIntron aiheutti väsymystä tai päänsärkyä noin kahdella kolmasosalla tutkittavista ja kuumetta tai jäykkyyttä noin puolella potilaista. Joidenkin näiden systeemisten oireiden (esim. kuume ja päänsärky) vakavuus väheni hoidon jatkuessa. Tutkimuksissa 1 ja 2 pistoskohdan tulehdusta ja reaktiota (esim. mustelmia, kutinaa ja ärsytystä) esiintyi noin kaksi kertaa enemmän PegIntron-hoitoa käytettäessä (jopa 75 %:lla koehenkilöistä) verrattuna INTRON A:han. Injektiokohdan kipu kuitenkin oli harvinainen (2-3 %) kaikissa ryhmissä. Tutkimuksessa 3 pistoskohdan reaktioiden tai tulehduksen yleinen ilmaantuvuus oli 23–24 %.
Potilaat, jotka saivat REBETOL/PegIntronia uusintahoitona aiemman interferoniyhdistelmähoidon epäonnistumisen jälkeen, raportoivat aiemmin hoitoa saamattomilla koehenkilöillä tehdyissä kliinisissä tutkimuksissa samanlaisia haittavaikutuksia kuin aiemmin tähän hoitoon liittyneet.
Pediatriset aiheet
Yleensä haittavaikutusprofiili lapsiväestössä oli samanlainen kuin aikuisilla. Lapsitutkimuksessa yleisimmät haittavaikutukset kaikilla koehenkilöillä olivat kuume (80 %), päänsärky (62 %), neutropenia (33 %), väsymys (30 %), anoreksia (29 %), pistoskohdan punoitus (29 %). %) ja oksentelua (27 %). Suurin osa tutkimuksessa raportoiduista haittavaikutuksista oli vaikeusasteeltaan lieviä tai kohtalaisia. Vakavia haittavaikutuksia raportoitiin 7 %:lla (8/107) kaikista koehenkilöistä, ja niihin kuuluivat pistoskohdan kipu (1 %), raajojen kipu (1 %), päänsärky (1 %), neutropenia (1 %) ja kuume (4 %). %). Tärkeitä haittavaikutuksia, joita esiintyi tässä tutkimusryhmässä, olivat hermostuneisuus (7 %; 7/107), aggressio (3 %; 3/107), viha (2 %; 2/107) ja masennus (1 %; 1/107) . Viisi koehenkilöä sai levotyroksiinihoitoa, kolme kliinistä kilpirauhasen vajaatoimintaa ja kahdella oireetonta TSH:n nousua. PegIntronilla ja REBETOLilla hoidettujen lapsipotilaiden painon ja pituuden nousu jäi normaaliväestötietojen ennustetusta jälkeen koko hoidon ajan. Vakavasti estynyt kasvunopeus (alle 3. prosenttipiste) havaittiin 70 %:lla koehenkilöistä hoidon aikana.
PegIntronin ja/tai ribaviriinin annosta jouduttiin muuttamaan 25 %:lla potilaista hoitoon liittyvien haittavaikutusten vuoksi, yleisimmin anemian, neutropenian ja painonpudotuksen vuoksi. Kaksi henkilöä (2 %; 2/107) keskeytti hoidon haittavaikutuksen vuoksi.
Haittavaikutukset, joiden ilmaantuvuus on suurempi tai yhtä suuri kuin 10 % lapsitutkimuksen koehenkilöillä, on esitetty taulukossa 7.
Yhdeksänkymmentäneljä 107 koehenkilöstä osallistui 5 vuoden pitkäaikaiseen seurantatutkimukseen. Pitkäaikaiset vaikutukset kasvuun olivat pienemmät niillä, joita hoidettiin 24 viikkoa, kuin niillä, joita hoidettiin 48 viikkoa. 24 prosentilla tutkittavista (11/46), joita hoidettiin 24 viikkoa, ja 40 prosentilla (19/48) 48 viikkoa hoidetuista koehenkilöistä (19/48) oli yli 15 prosenttipisteen lasku iän mukaan ennen hoitoa 5 vuoden loppuun. pitkäaikainen seuranta verrattuna hoitoa edeltäviin lähtötason prosenttipisteisiin. 11 prosentilla tutkimushenkilöistä (5/46), joita hoidettiin 24 viikkoa, ja 13 prosentilla (6/48) 48 viikkoa hoidetuista koehenkilöistä, havaittiin laskeneen hoitoa edeltäneestä lähtötasosta > 30 pituuden prosenttipistettä loppuun asti. 5 vuoden pitkäaikaisesta seurannasta. Vaikka havaittiin kaikissa ikäryhmissä, suurin riski pituuden alentumiseen pitkäaikaisen seurannan lopussa näytti korreloivan yhdistelmähoidon aloittamisen kanssa odotetun huippukasvun vuosien aikana. [Katso VAROITUKSET JA VAROTOIMET ]
Laboratorioarvot
Aikuiset ja lapsipotilaat
Haittavaikutusprofiili tutkimuksessa 3, jossa verrattiin PegIntron/painopohjaista REBETOL 200 mg yhdistelmää PegIntron/tasainen annos REBETOL 200 mg hoito-ohjelmaan, paljasti anemian lisääntyneen painoon perustuvalla annostuksella (29 % vs. 19 % painoon perustuvalla annostuksella). vs. tasainen annostusohjelma). Suurin osa anemiatapauksista oli kuitenkin lieviä ja reagoi annoksen pienentämiseen.
Valittujen laboratorioarvojen muutokset hoidon aikana yhdessä REBETOL 200 mg -hoidon kanssa on kuvattu alla. Hemoglobiinin, leukosyyttien, neutrofiilien ja verihiutaleiden väheneminen saattaa edellyttää annoksen pienentämistä tai hoidon pysyvää lopettamista [katso ANNOSTELU JA HALLINNOINTI ]. Muutokset valituissa laboratorioarvoissa hoidon aikana on kuvattu taulukossa 8. Suurin osa laboratorioarvojen muutoksista PegIntron/REBETOL 200 mg:n lapsitutkimuksessa oli lieviä tai kohtalaisia.
Hemoglobiini
Hemoglobiinitasot laskivat alle 11 g/dl noin 30 %:lla tutkimuksen 2 koehenkilöistä. Tutkimuksessa 3 47 %:lla tutkittavista, jotka saivat WBD REBETOL 200 mg ja 33 %:lla tasa-annos REBETOL 200 mg, hemoglobiinitasot laskivat alle 11 g. /dl. Hemoglobiinin alenemista alle 9 g/dl:iin esiintyi useammin WBD:tä saaneilla potilailla kuin tasa-annosteluilla (4 % ja 2 %). Tutkimuksessa 2 annoksen muuttamista tarvittiin 9 %:lla ja 13 %:lla potilaista PegIntron/REBETOL 200 mg ja INTRON A/REBETOL 200 mg ryhmissä. Tutkimuksessa 4 PegIntronia (1,5 mcg/kg)/REBETOLia saaneiden koehenkilöiden hemoglobiinitasot laskivat 8,5 - alle 10 g/dl (28 %) ja alle 8,5 g/dl (3 %), kun taas potilailla Pegasys 180 mikrog/Copegus saaneet nämä vähentymiset ilmenivät 26 %:lla ja 4 %:lla potilaista. Hemoglobiinitasot vakiintuivat keskimäärin hoitoviikoilla 4-6. Tyypillinen havaittu kuvio oli hemoglobiinitason lasku hoitoviikolla 4, jota seurasi stabiloituminen ja tasanne, joka säilyi hoidon loppuun asti. PegIntron-monoterapiatutkimuksessa hemoglobiinin lasku oli yleensä lievää ja annoksen muuttaminen oli harvoin tarpeen [ks. ANNOSTELU JA HALLINNOINTI ].
Neutrofiilit
Neutrofiilien määrän laskua havaittiin suurimmalla osalla aikuisista, joita hoidettiin yhdistelmähoidolla REBETOL 200 mg tutkimuksessa 2 (85 %) ja INTRON A/REBETOL (60 %). Vaikeaa mahdollisesti hengenvaarallista neutropeniaa (alle 0,5 x 109/l) esiintyi 2 %:lla INTRON A/REBETOL 200 mg:lla hoidetuista potilaista ja noin 4 %:lla potilaista, joita hoidettiin 200 mg PegIntron/REBETOL-annoksella tutkimuksessa 2. Kahdeksantoista prosentilla potilaista, jotka saivat tutkimuksessa 2 PegIntron/REBETOL vaati tutkimuksessa 2 interferonin annoksen muuttamista. Muutamat koehenkilöt (alle 1 %) vaativat hoidon pysyvän lopettamisen. Neutrofiilien määrät palautuivat yleensä hoitoa edeltäville tasoille 4 viikkoa hoidon lopettamisen jälkeen [ks. ANNOSTELU JA HALLINNOINTI ].
Verihiutaleet
Trombosyyttimäärät laskivat alle 100 000/mm³ noin 20 %:lla potilaista, joita hoidettiin pelkällä PegIntronilla tai REBETOL 200 mg:lla, ja 6 %:lla INTRON A/REBETOLilla hoidetuista aikuisista. Vakavaa verihiutaleiden määrän laskua (alle 50 000/mm³) esiintyy alle 4 %:lla aikuisista. Potilaat saattavat tarvita hoidon lopettamista tai annoksen muuttamista verihiutaleiden vähenemisen vuoksi [ks ANNOSTELU JA HALLINNOINTI ]. Tutkimuksessa 2 1 % tai 3 % koehenkilöistä tarvitsi INTRON A:n tai PegIntronin annosta muuttamaan vastaavasti. Verihiutalemäärät palautuivat yleensä hoitoa edeltävälle tasolle 4 viikkoa hoidon lopettamisen jälkeen.
Kilpirauhasen toiminta
TSH-poikkeavuuksien kehittyminen kliinisillä ilmenemismuodoilla tai ilman niitä liittyy interferonihoitoihin. Tutkimuksessa 2 kliinisesti ilmeisiä kilpirauhasen toimintahäiriöitä esiintyi henkilöillä, joita hoidettiin joko INTRON A:lla tai PegIntronilla (REBETOLin kanssa tai ilman) samankaltaisella ilmaantuvuusasteella (5 % kilpirauhasen vajaatoiminnasta ja 3 % kilpirauhasen liikatoiminnasta). Koehenkilöille kehittyi uusia TSH-poikkeavuuksia hoidon aikana ja seurantajakson aikana. Seurantajakson lopussa 7 %:lla koehenkilöistä oli edelleen epänormaaleja TSH-arvoja.
Bilirubiini ja virtsahappo
Tutkimuksessa 2 10-14 %:lle koehenkilöistä kehittyi hyperbilirubinemia ja 33-38 %:lle hyperurikemia hemolyysin yhteydessä. Kuudelle koehenkilölle kehittyi lievä tai kohtalainen kihti.
Kokemus kliinisistä kokeista – REBETOL/INTRON A -yhdistelmähoito
Aikuiset aiheet
Kliinisissä tutkimuksissa 19 % ja 6 % aiemmin hoitamattomista ja 6 % uusiutuneista potilaista keskeytti hoidon haittavaikutusten vuoksi yhdistelmähaaroissa, kun taas interferonihaaroissa vastaava luku oli 13 % ja 3 %. Valitut hoitoon liittyvät haittavaikutukset, joita esiintyi Yhdysvalloissa tehdyissä tutkimuksissa vähintään 5 %:n ilmaantuvuuden ollessa suurempi tai yhtä suuri kuin 5 %, on ilmoitettu hoitoryhmittäin (katso taulukko 9). Yleisesti ottaen valitut hoitoon liittyvät haittavaikutukset ilmoitettiin kansainvälisissä tutkimuksissa pienemmällä ilmaantuvuusasteella kuin Yhdysvalloissa, lukuun ottamatta asteniaa, influenssan kaltaisia oireita, hermostuneisuutta ja kutinaa.
Pediatriset aiheet
Kliinisissä tutkimuksissa, joihin osallistui 118 3–16-vuotiasta lasta, 6 % keskeytti hoidon haittavaikutusten vuoksi. Annosta jouduttiin muuttamaan 30 %:lla potilaista, yleisimmin anemian ja neutropenian vuoksi. Yleensä haittavaikutusprofiili lapsiväestössä oli samanlainen kuin aikuisilla. Pistoskohdan häiriöitä, kuumetta, ruokahaluttomuutta, oksentelua ja tunnelääkettä esiintyi useammin lapsipotilailla kuin aikuisilla. Sitä vastoin lapsipotilailla oli vähemmän väsymystä, dyspepsiaa, nivelsärkyä, unettomuutta, ärtyneisyyttä, keskittymishäiriöitä, hengenahdistusta ja kutinaa aikuisiin verrattuna. Valitut hoitoon liittyvät haittavaikutukset, joiden ilmaantuvuus oli suurempi tai yhtä suuri kuin 5 % kaikilla lapsilla, jotka saivat suositeltua REBETOL/INTRON A -yhdistelmähoitoannosta, on esitetty taulukossa 9.
48 viikon hoitojakson aikana havaittiin lineaarisen kasvun hidastuminen (keskimääräinen prosenttipisteen lasku 7 %) ja painon nousun nopeus (keskimääräinen prosenttipisteen lasku 9 %). Näiden suuntausten yleinen kääntyminen havaittiin 24 viikon hoidon jälkeisenä aikana. Pitkäaikaiset tiedot rajoitetulla määrällä potilaita viittaavat kuitenkin siihen, että yhdistelmähoito voi aiheuttaa kasvun eston, joka johtaa joidenkin potilaiden lopullisen aikuisen pituuden laskuun [ks. VAROITUKSET JA VAROTOIMET ].
Laboratorioarvot
Muutokset valikoiduissa hematologisissa arvoissa (hemoglobiini, valkosolut, neutrofiilit ja verihiutaleet) hoidon aikana on kuvattu alla (katso taulukko 10).
Hemoglobiini. Hemoglobiinin lasku REBETOL-hoitoa saaneiden potilaiden joukossa alkoi viikolla 1 ja vakiintui viikolla 4. Aiemmin hoitamattomilla potilailla, joita hoidettiin 48 viikkoa, keskimääräinen enimmäislasku lähtötasosta oli 3,1 g/dl Yhdysvaltain tutkimuksessa ja 2,9 g/dl kansainvälisessä tutkimuksessa. oikeudenkäyntiä. Relapsipotilailla keskimääräinen enimmäislasku lähtötasosta oli 2,8 g/dl Yhdysvalloissa ja 2,6 g/dl kansainvälisessä tutkimuksessa. Hemoglobiiniarvot palasivat hoitoa edeltäville tasoille 4–8 viikon kuluessa hoidon lopettamisesta useimmilla koehenkilöillä.
Bilirubiini ja virtsahappo. Kliinisissä tutkimuksissa havaittiin hemolyysiin liittyvää bilirubiinin ja virtsahapon nousua. Useimmat olivat kohtalaisia biokemiallisia muutoksia, ja ne hävisivät 4 viikon kuluessa hoidon lopettamisesta. Tämä havainto esiintyi useimmiten koehenkilöillä, joilla oli aiemmin diagnosoitu Gilbertin oireyhtymä. Tähän ei ole liitetty maksan toimintahäiriöitä tai kliinistä sairastuvuutta.
Markkinoinnin jälkeiset kokemukset
Seuraavat haittavaikutukset on tunnistettu ja raportoitu, kun REBETOL-valmistetta on käytetty yhdessä INTRON A:n tai PegIntronin kanssa hyväksynnän jälkeen. Koska nämä reaktiot on raportoitu vapaaehtoisesti epävarman kokoisesta populaatiosta, ei ole aina mahdollista luotettavasti arvioida niiden esiintymistiheyttä tai määrittää syy-yhteyttä lääkkeelle altistumiseen.
Veri- ja imukudosjärjestelmän häiriöt
Puhdas punasoluaplasia, aplastinen anemia
Korva- ja labyrinttihäiriöt
Kuulohäiriö, huimaus
Hengityselinten, rintakehän ja välikarsinan häiriöt
Keuhkoverenpainetauti
Silmäsairaudet
Seroottinen verkkokalvon irtauma
Endokriiniset häiriöt
Diabetes
HUUMEIDEN VUOROVAIKUTUKSET
Didanosiini
Altistuminen didanosiinille tai sen aktiiviselle metaboliitille (dideoksiadenosiini-5'-trifosfaatti) lisääntyy, kun didanosiinia annetaan samanaikaisesti ribaviriinin kanssa, mikä voi aiheuttaa tai pahentaa kliinisiä toksisuuksia. siksi REBETOL 200 mg kapseleiden tai oraaliliuoksen ja didanosiinin samanaikainen anto on vasta-aiheista. Kliinisissä tutkimuksissa on raportoitu kuolemaan johtaneita maksan vajaatoimintaa sekä perifeeristä neuropatiaa, haimatulehdusta ja oireista hyperlaktatemiaa/maitohappoasidoosia.
Nukleosidianalogit
Maksan vajaatoimintaa (jossain määrin kuolemaan johtanut) on ilmennyt maksakirroosipotilailla, joilla on samanaikainen HIV/HCV-infektio ja jotka ovat saaneet antiretroviraalista yhdistelmähoitoa HIV:n ja interferoni alfan ja ribaviriinin vuoksi. Alfa-interferonihoidon lisääminen yksinään tai yhdessä ribaviriinin kanssa voi lisätä riskiä tässä potilasryhmässä. Potilaita, jotka saavat interferonia ribaviriinin ja nukleosidisten käänteiskopioijaentsyymin estäjien (NRTI) kanssa, tulee seurata tarkasti hoitoon liittyvien toksisuusvaikutusten, erityisesti maksan vajaatoiminnan ja anemian varalta. NRTI-lääkityksen lopettamista tulee pitää lääketieteellisesti tarkoituksenmukaisena (ks yksittäisen NRTI-tuotteen merkinnät ). Interferonin, ribaviriinin tai molempien annoksen pienentämistä tai lopettamista tulee myös harkita, jos havaitaan kliinisen toksisuuden pahenemista, mukaan lukien maksan vajaatoiminta (esim. Child-Pugh yli 6).
Ribaviriini saattaa antagonisoida stavudiinin ja tsidovudiinin soluviljelmän antiviraalista aktiivisuutta HIV:tä vastaan. Ribaviriinin on osoitettu soluviljelmässä estävän lamivudiinin, stavudiinin ja tsidovudiinin fosforylaatiota, mikä saattaa heikentää antiretroviraalista aktiivisuutta. Toisella pegyloidulla interferonilla yhdessä ribaviriinin kanssa tehdyssä tutkimuksessa ei kuitenkaan havaittu farmakokineettisiä (esim. plasmapitoisuudet tai solunsisäiset trifosforyloitujen aktiivisten metaboliittien pitoisuudet) tai farmakodynaamisia (esim. HIV/HCV:n virologisen suppression menetys) yhteisvaikutuksia, kun ribaviriini ja lamivudiini (n) = 18), stavudiinia (n = 10) tai tsidovudiinia (n = 6) annettiin samanaikaisesti osana usean lääkkeen hoitoa HIV/HCV-infektoituneille potilaille. Siksi ribaviriinin samanaikaisessa käytössä jommankumman näistä lääkkeistä tulee noudattaa varovaisuutta.
Sytokromi P-450:n metaboloimat lääkkeet
In vitro -tutkimusten tulokset, joissa käytettiin sekä ihmisen että rotan maksan mikrosomivalmisteita, osoittivat vain vähän tai ei ollenkaan sytokromi P-450 -entsyymivälitteistä ribaviriinin metaboliaa, ja P-450-entsyymipohjaisten lääkeinteraktioiden mahdollisuus oli minimaalinen.
INTRON A:n ja REBETOL 200 mg:n kapseleiden välillä ei havaittu farmakokineettisiä yhteisvaikutuksia usean annoksen farmakokineettisessä tutkimuksessa.
Atsatiopriini
Ribaviriinin käytön kroonisen hepatiitti C:n hoitoon atsatiopriinia saavilla potilailla on raportoitu aiheuttavan vaikeaa pansytopeniaa ja saattaa lisätä atsatiopriiniin liittyvän myelotoksisuuden riskiä. Inosiinimonofosfaattidehydrogenaasia (IMDH) tarvitaan yhdelle atsatiopriinin metaboliareiteistä. Ribaviriinin tiedetään estävän IMDH:ta, mikä johtaa atsatiopriinimetaboliitin, 6-metyylitioinosiinimonofosfaatin (6-MTITP) kertymiseen, mikä liittyy myelotoksisuuteen (neutropenia, trombosytopenia ja anemia). Atsatiopriinia ribaviriinin kanssa saavien potilaiden täydellinen verenkuva, mukaan lukien verihiutaleiden määrä, tulee seurata viikoittain ensimmäisen kuukauden ajan, kahdesti kuukaudessa toisen ja kolmannen hoitokuukauden ajan, sitten kuukausittain tai useammin, jos annostusta tai muita hoitomuutoksia tarvitaan [ks. VAROITUKSET JA VAROTOIMET ].
VAROITUKSET
Mukana osana VAROTOIMENPITEET osio.
VAROTOIMENPITEET
Raskaus
REBETOL 200 mg kapselit ja oraaliliuos voivat aiheuttaa sikiövaurioita ja syntymättömän lapsen kuoleman. REBETOL-hoitoa ei saa aloittaa ennen kuin on saatu negatiivinen raskaustesti juuri ennen suunniteltua hoidon aloittamista. Potilaiden tulee käyttää vähintään kahta ehkäisymenetelmää ja käydä kuukausittain raskaustesteillä hoidon aikana ja 6 kuukauden ajan hoidon lopettamisen jälkeen. Naispotilaiden ja miespotilaiden naispuolisten kumppanien raskauden välttämiseksi on oltava erittäin varovainen. REBETOL 200mg on osoittanut merkittäviä teratogeenisia ja alkioita tappavia vaikutuksia kaikissa eläinlajeissa, joilla on suoritettu riittävät tutkimukset. Nämä vaikutukset ilmenivät annoksilla kuten alhainen kuin yksi kahdeskymmenesosa ihmisille suositellusta ribaviriiniannoksesta. REBETOL 200 mg -hoitoa ei saa aloittaa ennen kuin raskaustesti on negatiivinen on saatu välittömästi ennen suunniteltua hoidon aloittamista [katso LAATIKKO VAROITUS , VASTA-AIHEET , Käyttö tietyissä populaatioissa , ja POTILASTIEDOT ].
Anemia
Ribaviriinin ensisijainen toksisuus on hemolyyttinen anemia, jota havaittiin kliinisissä tutkimuksissa noin 10 %:lla REBETOL/INTRON A:lla hoidetuista potilaista. REBETOL-kapseleihin liittyvä anemia ilmaantuu 1-2 viikon kuluessa hoidon aloittamisesta. Koska hemoglobiinin alkupudotus voi olla merkittävä, on suositeltavaa mitata hemoglobiini tai hematokriitti ennen hoidon aloittamista sekä hoidon viikolla 2 ja 4 tai useammin, jos se on kliinisesti aiheellista. Potilaita tulee sitten seurata kliinisesti tarkoituksenmukaisella tavalla [katso ANNOSTELU JA HALLINNOINTI ].
Kuolemaan johtaneita ja ei-fataalisia sydäninfarkteja on raportoitu potilailla, joilla on REBETOLin aiheuttama anemia. Ennen ribaviriinihoidon aloittamista potilailla on arvioitava sydänsairaus. Potilaille, joilla on aiempi sydänsairaus, tulee ottaa EKG-kuvaus ennen hoitoa, ja heitä on seurattava asianmukaisesti hoidon aikana. Jos kardiovaskulaarinen tila heikkenee, hoito on keskeytettävä tai lopetettava [ks. ANNOSTELU JA HALLINNOINTI ]. Koska lääkkeiden aiheuttama anemia voi pahentaa sydänsairautta, potilaiden, joilla on ollut merkittävä tai epästabiili sydänsairaus, ei tule käyttää REBETOLia.
Haimatulehdus
REBETOL- ja INTRON A- tai PegIntron-hoito tulee keskeyttää potilailla, joilla on haimatulehduksen merkkejä ja oireita, ja lopettaa potilaiden, joilla on todettu haimatulehdus.

Keuhkosairaudet
Keuhkooireita, mukaan lukien hengenahdistus, keuhkoinfiltraatit, keuhkotulehdus, keuhkoverenpainetauti ja keuhkokuume, on raportoitu REBETOL-hoidon aikana alfa-interferoniyhdistelmähoidon kanssa. ajoittain kuolemaan johtaneita keuhkokuumeita on esiintynyt. Lisäksi sarkoidoosia tai sarkoidoosin pahenemista on raportoitu. Jos on näyttöä keuhkoinfiltraateista tai keuhkojen toiminnan heikkenemisestä, potilasta on seurattava tarkasti ja tarvittaessa yhdistelmähoito on lopetettava.
Oftalmologiset häiriöt
Ribaviriinia käytetään yhdistelmähoidossa alfa-interferonien kanssa. Alfa-interferonihoito aiheuttaa tai pahentaa näön heikkenemistä tai menetystä, retinopatiaa, mukaan lukien silmänpohjan turvotus, verkkokalvon valtimo tai laskimo, tromboosi, verkkokalvon verenvuoto ja vanutäplät, optinen neuriitti, papillideema ja seroosin verkkokalvon irtauma. Kaikille potilaille on tehtävä silmätutkimus lähtötilanteessa. Potilaille, joilla on aiempaa silmäsairauksia (esim. diabeettinen tai hypertensiivinen retinopatia), tulee käydä säännöllisesti silmätutkimuksissa alfa-interferonihoidon yhdistelmähoidon aikana. Jokaisen potilaan, jolle kehittyy silmäoireita, tulee saada nopea ja täydellinen silmätutkimus. Alfa-interferonien yhdistelmähoito tulee lopettaa potilailla, joille kehittyy uusia tai pahenevia silmäsairauksia.
Laboratoriotestit
PegIntron yhdessä ribaviriinin kanssa voi aiheuttaa vakavaa neutrofiilien ja verihiutaleiden määrän laskua sekä hematologisia, endokriinisiä (esim. TSH) ja maksan poikkeavuuksia.
PegIntron/REBETOL-yhdistelmähoitoa saaville potilaille tulee tehdä hematologiset ja verikemialliset testit ennen hoidon aloittamista ja sen jälkeen määräajoin. Aikuisten kliinisessä tutkimuksessa mitattiin täydellinen verenkuva (mukaan lukien hemoglobiini-, neutrofiilien ja verihiutaleiden määrä) ja kemiat (mukaan lukien ASAT, ALAT, bilirubiini ja virtsahappo) hoitojakson aikana viikoilla 2, 4, 8, 12 ja sitten 6 viikon välein tai useammin, jos poikkeavuuksia kehittyy. Lapsipotilailla samat laboratorioparametrit arvioitiin hemoglobiinin lisäarvioinnilla hoitoviikolla 6. TSH-tasot mitattiin 12 viikon välein hoitojakson aikana. HCV-RNA tulee mitata säännöllisesti hoidon aikana [katso ANNOSTELU JA HALLINNOINTI ].
Hammas- ja periodontaaliset sairaudet
Hammas- ja periodontaalisia häiriöitä on raportoitu potilailla, jotka ovat saaneet ribaviriinin ja interferonin tai peginterferonin yhdistelmähoitoa. Lisäksi suun kuivuminen voi vaikuttaa hampaisiin ja suun limakalvoihin pitkäaikaisen hoidon aikana REBETOLin ja pegyloidun tai pegyloidun interferoni alfa-2b:n yhdistelmällä. Potilaiden tulee harjata hampaat perusteellisesti kahdesti päivässä ja käydä säännöllisissä hammastutkimuksissa. Jos oksentaa, heitä tulee neuvoa huuhtelemaan suunsa huolellisesti sen jälkeen.
Atsatiopriinin samanaikainen anto
Pansytopeniaa (huomattava punasolujen, neutrofiilien ja verihiutaleiden väheneminen) ja luuytimen suppressiota on kirjallisuudessa raportoitu esiintyvän 3–7 viikon kuluessa pegyloidun interferonin/ribaviriinin ja atsatiopriinin samanaikaisesta annosta. Tällä rajoitetulla potilaiden määrällä (n=8) myelotoksisuus korjaantui 4–6 viikossa, kun sekä HCV:n antiviraalinen hoito että samanaikainen atsatiopriinihoito lopetettiin, eikä se uusiutunut, kun kumpikaan hoito aloitettiin uudelleen yksinään. PegIntronin, REBETOLin ja atsatiopriinin käyttö tulee lopettaa pansytopenian vuoksi, eikä pegyloitua interferonia/ribaviriinia tule ottaa uudelleen käyttöön samanaikaisen atsatiopriinin kanssa (ks. HUUMEIDEN VUOROVAIKUTUKSET ].
Vaikutus kasvuun – Käyttö lapsille
Tiedot PegIntronin ja REBETOLin vaikutuksista kasvuun ovat peräisin 3–17-vuotiailla koehenkilöillä tehdystä avoimesta tutkimuksesta, jossa painon ja pituuden muutoksia verrataan Yhdysvaltojen normatiivisiin väestötietoihin. Yleensä PegIntronilla ja REBETOL 200 mg:lla hoidettujen lasten painon ja pituuden nousu on jäljessä normaaliväestötietojen ennusteesta koko hoidon ajan. Vakavasti estynyt kasvunopeus (alle 3. prosenttipiste) havaittiin 70 %:lla koehenkilöistä hoidon aikana. Hoidon jälkeen useimmilla koehenkilöillä esiintyi rebound-kasvua ja painonnousua. Pitkäaikaiset seurantatiedot lapsipotilaista osoittavat kuitenkin, että PegIntron yhdistelmähoidossa REBETOLin kanssa saattaa aiheuttaa kasvun estymistä, mikä johtaa aikuisen pituuden laskuun joillakin potilailla [ks. HAITTAVAIKUTUKSET ].
Vastaavasti vaikutusta kasvuun havaittiin koehenkilöillä yhden vuoden kestäneen REBETOL 200 mg:n ja INTRON A:n yhdistelmähoidon jälkeen. Pitkäaikaisessa seurantatutkimuksessa rajoitetulla määrällä näitä koehenkilöitä yhdistelmähoito johti aikuisen lopullisen pituuden laskuun joillakin koehenkilöillä [ks. HAITTAVAIKUTUKSET ].
Käyttösuojat
Kliinisten tutkimusten tulosten perusteella ribaviriinimonoterapia ei ole tehokas kroonisen hepatiitti C -virusinfektion hoidossa. siksi REBETOL 200 mg kapseleita tai oraaliliuosta ei saa käyttää yksinään. REBETOL-kapseleiden ja oraaliliuoksen turvallisuus ja teho on osoitettu vain käytettäessä niitä yhdessä INTRON A:n tai PegIntronin (ei muiden interferonien) kanssa yhdistelmähoitona.
REBETOL/INTRON A- ja PegIntron-hoidon turvallisuutta ja tehoa HIV-infektion, adenovirus-, RSV-, parainfluenssa- tai influenssainfektioiden hoidossa ei ole osoitettu. REBETOL 200 mg kapseleita ei tule käyttää näihin käyttöaiheisiin. Inhaloitavaksi tarkoitettu ribaviriini on varustettu erillisellä merkinnällä, jota tulee kuulla, jos harkitaan ribaviriinin inhalaatiohoitoa.
REBETOL/INTRON A- tai PegIntron-hoito aiheuttaa merkittäviä haittavaikutuksia, mukaan lukien vaikea masennus ja itsemurha-ajatukset, hemolyyttinen anemia, luuytimen toiminnan heikkeneminen, autoimmuuni- ja infektiosairaudet, keuhkojen toimintahäiriöt, haimatulehdus ja diabetes. Itsemurha-ajatuksia tai -yrityksiä esiintyi useammin lapsipotilailla, pääasiassa nuorilla, aikuisiin verrattuna (2,4 % vs. 1 %) hoidon ja hoidon ulkopuolisen seurannan aikana. INTRON A:n ja PegIntronin merkinnät tulee tarkistaa kokonaisuudessaan lisäturvallisuustietojen saamiseksi ennen yhdistelmähoidon aloittamista.
Potilasneuvontatiedot
Neuvo potilasta lukemaan FDA:n hyväksymä potilasmerkintä ( Lääkitysopas ).
Anemia
Yleisin REBETOL-kapseleiden haittavaikutus on anemia, joka voi olla vakava [katso VAROITUKSET JA VAROTOIMET ja HAITTAVAIKUTUKSET ]. Potilaita tulee kertoa, että laboratoriotutkimukset ovat tarpeen ennen hoidon aloittamista ja ajoittain sen jälkeen [ks. ANNOSTELU JA HALLINNOINTI ]. On suositeltavaa, että potilaat ovat hyvin nesteytettyinä, erityisesti hoidon alkuvaiheessa.
Raskaus
Potilaille on kerrottava, että REBETOL 200 mg kapselit ja oraaliliuos voivat aiheuttaa synnynnäisiä epämuodostumia ja syntymättömän lapsen kuoleman. REBETOL 200mg -valmistetta eivät saa käyttää raskaana olevat naiset tai miehet, joiden naispuoliset kumppanit ovat raskaana. Äärimmäistä varovaisuutta on noudatettava raskauden välttämiseksi naispotilailla ja REBETOL-hoitoa saavien miespotilaiden naispuolisoilla. REBETOL-hoitoa ei saa aloittaa ennen kuin on saatu raportti negatiivisesta raskaustestistä välittömästi ennen hoidon aloittamista. Potilaiden on tehtävä raskaustesti kuukausittain hoidon aikana ja 6 kuukauden ajan hoidon jälkeen.
Hedelmällisessä iässä olevia naisia tulee neuvoa tehokkaan ehkäisyn (kaksi luotettavaa muotoa) käytöstä ennen hoidon aloittamista. Potilaita (miehiä ja naisia) on kerrottava teratogeenisistä/sikiövaurioista ja heitä on neuvottava käyttämään tehokasta ehkäisyä REBETOL 200 mg -annoksen aikana ja 6 kuukauden ajan hoidon jälkeen. Potilaita (miehiä ja naisia) tulee neuvoa ilmoittamaan lääkärille välittömästi, jos he tulevat raskaaksi [katso VASTA-AIHEET , VAROITUKSET JA VAROTOIMET , ja Käyttö tietyissä populaatioissa ].
Jos raskaus tulee hoidon aikana tai 6 kuukautta hoidon jälkeen, potilasta on kerrottava REBETOL 200 mg -hoidon teratogeenisestä riskistä sikiölle. Potilaiden tai potilaiden kumppanien tulee välittömästi ilmoittaa lääkärilleen raskaudesta, joka ilmenee hoidon aikana tai 6 kuukauden sisällä hoidon lopettamisesta. Lääkkeen määrääjien tulee ilmoittaa tällaisista tapauksista soittamalla numeroon 1-800-593-2214.
Riskit vs. edut
REBETOL-kapseleita saaville potilaille tulee kertoa hoidon eduista ja riskeistä, ohjata sen asianmukaista käyttöä ja ohjata potilaalle Lääkitysopas . Potilaille tulee kertoa, että hepatiitti C -infektion hoidon vaikutusta tartuntatautiin ei tunneta, ja että asianmukaisia varotoimia on ryhdyttävä hepatiitti C -viruksen leviämisen estämiseksi.
Potilaille tulee kertoa, mitä tehdä, jos he unohtavat ottaa REBETOL-annoksen. unohtunut annos tulee ottaa mahdollisimman pian saman päivän aikana. Potilaat eivät saa kaksinkertaistaa seuraavaa annosta. Potilaita tulee neuvoa ottamaan yhteyttä terveydenhuollon tarjoajaan, jos heillä on kysyttävää.
Ei-kliininen toksikologia
Karsinogeneesi, mutageneesi, hedelmällisyyden heikkeneminen
Karsinogeneesi
Ribaviriini ei aiheuttanut minkään kasvaintyypin lisääntymistä, kun sitä annettiin kuuden kuukauden ajan siirtogeeniselle p53-puutteelliselle hiirimallille annoksilla 300 mg/kg asti (arvioitu ihmisarvo 25 mg/kg perustuen kehon pinta-alan säätöön 60 kg painavalla aikuisella ; noin 1,9 kertaa ihmisen suurin suositeltu päiväannos). Ribaviriini ei ollut syöpää aiheuttava, kun sitä annettiin rotille 2 vuoden ajan annoksilla 40 mg/kg asti (arvioitu ihmisarvo 5,71 mg/kg perustuen kehon pinta-alan säätöön 60 kg painavalla aikuisella).
Mutageneesi
Ribaviriini osoitti lisääntynyttä mutaatioiden ja solutransformaatioiden ilmaantuvuutta useissa genotoksisuusmäärityksissä. Ribaviriini oli aktiivinen Balb/3T3 in vitro -solutransformaatiomäärityksessä. Mutageeninen aktiivisuus havaittiin hiiren lymfoomamäärityksessä ja annoksilla 20-200 mg/kg (arvioitu ihmisen ekvivalentti 1,67-16,7 mg/kg, perustuen kehon pinta-alan säätöön 60 kg painavalla aikuisella; 0,1-1 kertaa maksimi ihmisen suositeltu ribaviriinin 24 tunnin annos) hiiren mikrotumatestissä. Hallitseva tappava määritys rotilla oli negatiivinen, mikä osoitti, että jos rotissa tapahtui mutaatioita, ne eivät välittyneet urospuolisten sukusolujen kautta.
Hedelmällisyyden heikkeneminen
Ribaviriini osoitti merkittäviä alkioita tappavia ja teratogeenisiä vaikutuksia annoksilla, jotka ovat selvästi alle suositeltua ihmisannostusta kaikissa eläinlajeissa, joilla on suoritettu riittävät tutkimukset. Kallon, kitalaen, silmän, leuan, raajojen, luuston ja maha-suolikanavan epämuodostumia havaittiin. Teratogeenisten vaikutusten ilmaantuvuus ja vakavuus lisääntyivät lääkeannoksen noustessa. Sikiöiden ja jälkeläisten eloonjääminen väheni. Perinteisissä sikiötoksisuus/teratogeenisuustutkimuksissa rotilla ja kaniineilla havaitut vaikutuksettomat annostasot olivat selvästi alle ehdotetun kliinisen käytön (0,3 mg/kg/vrk sekä rotalla että kaniinilla; noin 0,06 kertaa suositeltu 24 tunnin annos ihmiselle) ribaviriini). Peri/postnataalisessa toksisuustutkimuksessa rotilla ei havaittu emolle toksisuutta tai vaikutuksia jälkeläisiin suun kautta enintään 1 mg/kg/vrk (arvioitu ihmisen ekvivalenttiannos 0,17 mg/kg perustuen kehon pinta-alan säätöön 60 kg painoisella aikuisella) ; noin 0,01 kertaa ihmisen suurin suositeltu ribaviriinin 24 tunnin annos) [ks. VASTA-AIHEET , ja VAROITUKSET JA VAROTOIMET ].
Hedelmälliset naiset ja hedelmällisten naisten kumppanit eivät saa saada REBETOLia, elleivät potilas ja hänen kumppaninsa käytä tehokasta ehkäisyä (kaksi luotettavaa muotoa). Koska ribaviriinin usean annoksen puoliintumisaika (t1/2) on 12 päivää, tehokasta ehkäisyä on käytettävä 6 kuukauden ajan hoidon jälkeen (esim. ribaviriinin puhdistuman puoliintumisaika on 15).
REBETOLia tulee käyttää varoen hedelmällisillä miehillä. Hiirillä tehdyissä tutkimuksissa, joissa arvioitiin ribaviriinin aiheuttaman kivesten rappeutumisen ajan kulumista ja palautuvuutta annoksilla 15-150 mg/kg/vrk (arvioitu ihmisen vastaava 1,25-12,5 mg/kg/vrk, perustuen kehon pinta-alan säätöön 60-kg aikuinen; 0,1-0,8 kertaa ribaviriinin enimmäisannos ihmiselle 24 tunnin ajan) annettuna 3 tai 6 kuukauden ajan, siittiöissä esiintyi poikkeavuuksia. Hoidon lopettamisen jälkeen ribaviriinin aiheuttamasta kivestoksisuudesta toipuminen oli ilmeistä 1 tai 2 spermatogeneesisyklin aikana.
Käyttö tietyissä populaatioissa
Raskaus
Raskausluokka X
[Katso VASTA-AIHEET , VAROITUKSET JA VAROTOIMET , ja Ei-kliininen toksikologia ].
Hoito ja jälkihoito:
Mahdollinen riski sikiölle
Ribaviriinin tiedetään kerääntyvän solunsisäisiin komponentteihin, joista se poistuu hyvin hitaasti. Ei tiedetä, onko siittiöiden sisältämällä ribaviriinilla mahdollinen teratogeeninen vaikutus munasolujen hedelmöityksessä. Rotilla tehdyssä tutkimuksessa pääteltiin, että ribaviriini ei indusoinut hallitsevaa kuolleisuutta annoksilla 200 mg/kg 5 päivän ajan (arvioidut ihmisen vastaavat annokset 7,14–28,6 mg/kg, perustuen kehon pinta-alan säätöön 60 kg aikuisen; jopa 1,7 kertaa ihmisen suurin suositeltu ribaviriiniannos). Ribaviriinin mahdollisten ihmisiin kohdistuvien teratogeenisten vaikutusten vuoksi miespotilaita tulee kuitenkin neuvoa ryhtymään kaikkiin varotoimiin naispuolisten kumppanien raskauden riskin välttämiseksi.
Hedelmällisessä iässä olevat naiset eivät saa saada REBETOLia, elleivät he käytä tehokasta ehkäisyä (kaksi luotettavaa muotoa) hoidon aikana. Lisäksi tehokasta ehkäisyä tulee käyttää 6 kuukauden ajan hoidon jälkeen, koska ribaviriinin toistuvan annoksen puoliintumisaika (t1/2) on 12 päivää.
Miespotilaiden ja heidän naiskumppaniensa on käytettävä tehokasta ehkäisyä (kaksi luotettavaa muotoa) REBETOL-hoidon aikana ja 6 kuukauden hoidon jälkeisenä aikana (esim. 15 puoliintumisaikaa ribaviriinin poistumiselle kehosta).
Ribaviriinin raskausrekisteri on perustettu seuraamaan raskauksien äidin ja sikiön tuloksia naispotilailla ja miespotilaiden naispuolisilla kumppaneilla, jotka ovat altistuneet ribaviriinille hoidon aikana ja 6 kuukauden ajan hoidon lopettamisen jälkeen. Lääkäreitä ja potilaita kehotetaan ilmoittamaan tällaisista tapauksista soittamalla numeroon 1-800-593-2214.
Imettävät äidit
Ei tiedetä, erittyykö REBETOL-valmiste äidinmaitoon. Koska lääke voi aiheuttaa vakavia haittavaikutuksia imeväisille, on tehtävä päätös, lopetetaanko imetys vai lykätäänkö tai lopetetaanko REBETOL-hoito.
Käyttö lapsille
REBETOL 200 mg:n turvallisuutta ja tehoa yhdessä PegIntronin kanssa ei ole osoitettu alle 3-vuotiailla lapsipotilailla. REBETOL/INTRON A -hoidossa on otettava huomioon sairauden etenemisen todisteet, kuten maksatulehdus ja fibroosi, sekä vasteen ennustetekijät, HCV-genotyyppi ja viruskuorma, kun lapsipotilaan hoitoa päätetään. Hoidon hyötyjä tulee punnita havaittuja turvallisuushavaintoja vastaan.
Pitkäaikaiset seurantatiedot lapsipotilaista osoittavat, että REBETOL yhdessä PegIntronin tai INTRON A:n kanssa saattaa aiheuttaa kasvun estymistä, mikä johtaa joidenkin potilaiden pituuden laskuun [ks. VAROITUKSET JA VAROTOIMET ja HAITTAVAIKUTUKSET ].
Itsemurha-ajatuksia tai -yrityksiä esiintyi useammin lapsipotilailla, pääasiassa nuorilla, verrattuna aikuisiin potilaisiin (2,4 % vs. 1 %) hoidon ja hoidon ulkopuolisen seurannan aikana [katso VAROITUKSET JA VAROTOIMET ]. Kuten aikuisilla potilailla, lapsipotilailla oli muita psykiatriset haittavaikutukset (esim. masennus, emotionaalinen labilisuus, uneliaisuus), anemia ja neutropenia [ks. VAROITUKSET JA VAROTOIMET ].
Geriatrinen käyttö
REBETOL/INTRON A- tai PegIntron-hoidon kliinisissä tutkimuksissa ei ollut riittävästi 65-vuotiaita ja sitä vanhempia koehenkilöitä sen määrittämiseksi, reagoivatko he eri tavalla kuin nuoremmat.
REBETOL 200 mg:n tiedetään erittyvän olennaisesti munuaisten kautta, ja tämän lääkkeen toksisten reaktioiden riski voi olla suurempi potilailla, joilla on heikentynyt munuaisten toiminta. Koska iäkkäillä potilailla on usein heikentynyt munuaisten toiminta, annoksen valinnassa on oltava varovainen. Munuaisten toimintaa on seurattava ja annosta on muutettava vastaavasti. REBETOL 200 mg:aa ei tule käyttää potilailla, joiden kreatiniinipuhdistuma on alle 50 ml/min [ks. VASTA-AIHEET ].
Yleensä REBETOL 200 mg kapseleita tulee antaa iäkkäille potilaille varoen, alkaen annosalueen alemmasta päästä, mikä kuvastaa maksan ja sydämen toiminnan heikkenemisen sekä samanaikaisen sairauden tai muun lääkehoidon yleisyyttä. Kliinisissä tutkimuksissa iäkkäillä koehenkilöillä oli yleisempi anemia (67 %) kuin nuoremmilla potilailla (28 %) [ks. VAROITUKSET JA VAROTOIMET ].
Elinsiirtojen vastaanottajat
INTRON A:n ja PegIntronin turvallisuutta ja tehoa yksinään tai yhdessä REBETOLin kanssa hepatiitti C:n hoidossa maksan tai muiden elinsiirtojen saajilla ei ole varmistettu. Pienessä (n=16) yhden keskuksen kontrolloimattomassa tapauskokemuksessa munuaisten vajaatoiminta alfa-interferonin ja ribaviriinin yhdistelmähoitoa saavilla munuaissiirteen saajilla oli yleisempää kuin odotettiin keskuksen aiemman kokemuksen perusteella munuaissiirteen saajilla, jotka eivät saaneet yhdistelmähoitoa. Munuaisten vajaatoiminnan suhde munuaissiirteen hylkimiseen ei ole selvä.
HIV- tai HBV-yhteisinfektio
PegIntron/REBETOL 200 mg:n ja INTRON A/REBETOLin turvallisuutta ja tehoa hoidettaessa potilaita, joilla on HCV-infektio, joilla on samanaikaisesti HIV- tai HBV-infektio, ei ole varmistettu.
YLIANNOSTUS
Yliannostuksesta on vain vähän kokemusta. Enintään 20 g:n REBETOL-kapseleiden, INTRON A:n 120 miljoonan yksikön akuutin nielemisen ja INTRON A:n ihonalaisia annoksia, jotka ovat jopa 10 kertaa suositeltuja annoksia suurempia, on raportoitu. Ensisijaisia havaittuja vaikutuksia ovat INTRON A:n ja REBETOLin terapeuttiseen käyttöön liittyvien haittavaikutusten lisääntynyt esiintyvyys ja vakavuus. Maksaentsyymihäiriöitä, munuaisten vajaatoimintaa, verenvuotoa ja sydäninfarktia on kuitenkin raportoitu, kun INTRON A:ta on annettu ihonalaisesti kerta-annoksina, jotka ylittävät annossuositukset.
INTRON A:n tai REBETOL 200 mg:n yliannostukselle ei ole spesifistä vastalääkettä, ja hemodialyysi ja peritoneaalidialyysi eivät ole tehokkaita näiden aineiden yliannostuksen hoidossa.
VASTA-AIHEET
REBETOL-yhdistelmähoito on vasta-aiheinen:
- raskaana olevat naiset. REBETOL 200 mg voi aiheuttaa sikiövaurioita, kun sitä annetaan raskaana olevalle naiselle. REBETOL on vasta-aiheinen naisille, jotka ovat tai voivat tulla raskaaksi. Jos REBETOLia käytetään raskauden aikana tai jos potilas tulee raskaaksi käyttäessään REBETOL 200 mg -valmistetta, potilasta on informoitava mahdollisesta vaarasta sikiölle [ks. VAROITUKSET JA VAROTOIMET , Käyttö tietyissä populaatioissa , ja POTILASTIEDOT ]
- miehet, joiden naispuoliset kumppanit ovat raskaana
- potilaat, joilla on tunnettuja yliherkkyysreaktioita, kuten Stevens-Johnsonin oireyhtymä, toksinen, epidermaalinen nekrolyysi ja erythema multiforme ribaviriinille tai jollekin valmisteen aineosalle
- potilaille, joilla on autoimmuunihepatiitti
- potilaat, joilla on hemoglobinopatia (esim. suuri talassemia, sirppisoluanemia)
- potilailla, joiden kreatiniinipuhdistuma on alle 50 ml/min. [katso Käyttö tietyissä populaatioissa ja KLIININEN FARMAKOLOGIA ]
- REBETOLin ja didanosiinin samanaikainen käyttö on vasta-aiheista, koska altistuminen didanosiinin aktiiviselle metaboliitille (dideoksiadenosiini-5'-trifosfaatti) lisääntyy. Kuolemaan johtavaa maksan vajaatoimintaa sekä perifeeristä neuropatiaa, haimatulehdusta ja oireista hyperlaktatemiaa/maitohappoasidoosia on raportoitu potilailla, jotka ovat saaneet didanosiinia yhdessä ribaviriinin kanssa [ks. HUUMEIDEN VUOROVAIKUTUKSET ].
KLIININEN FARMAKOLOGIA
Toimintamekanismi
Ribaviriini on viruslääke [katso Mikrobiologia ].
Farmakokinetiikka
Taulukossa 11 on yhteenveto kerta- ja toistuvan annoksen farmakokineettisistä ominaisuuksista aikuisilla. Ribaviriini imeytyi nopeasti ja laajasti suun kautta annetun annon jälkeen. Ensikierron metabolian vuoksi absoluuttinen hyötyosuus oli kuitenkin keskimäärin 64 % (44 %). Annoksen ja AUCtf:n (AUC ajankohdasta nollasta viimeiseen mitattavissa olevaan pitoisuuteen) välillä oli lineaarinen suhde 200–1200 mg ribaviriinin kerta-annoksilla. Annoksen ja Cmax:n välinen suhde oli käyräviivainen, ja se oli yleensä oireeton yli 400–600 mg:n kerta-annoksia.
Toistuvan oraalisen annostelun yhteydessä havaittiin ribaviriinin 6-kertainen kertyminen plasmassa AUC12h:n perusteella. Kun annettiin suun kautta 600 mg kahdesti vuorokaudessa, vakaa tila saavutettiin noin 4 viikossa, ja keskimääräiset vakaan tilan pitoisuudet plasmassa olivat 2200 ng/ml (37 %). Annostelun lopettamisen jälkeen keskimääräinen puoliintumisaika oli 298 (30 %) tuntia, mikä todennäköisesti heijastaa hidasta eliminaatiota ei-plasmaosastoista.
Antasidin vaikutus ribaviriinin imeytymiseen
REBETOL-kapseleiden samanaikainen antaminen magnesiumia, alumiinia ja simetikonia sisältävän antasidin kanssa johti 14 %:n laskuun ribaviriinin keskimääräisessä AUCtf:ssä. Tämän kerta-annostutkimuksen tulosten kliinistä merkitystä ei tunneta.
Kudosten jakautuminen
Ribaviriinin kuljetusta ei-plasmaosastoihin on tutkittu laajimmin punasoluissa, ja sen on todettu tapahtuvan ensisijaisesti es-tyypin tasapainoisen nukleosidikuljettajan kautta. Tämän tyyppinen kuljettaja on läsnä käytännöllisesti katsoen kaikissa solutyypeissä ja voi selittää laajan jakautumistilavuuden. Ribaviriini ei sitoudu plasman proteiineihin.
Aineenvaihdunta ja erittyminen
Ribaviriinilla on kaksi metaboliareittiä: (i) palautuva fosforylaatioreitti tumallisissa soluissa; ja (ii) hajoamisreitti, johon liittyy deribosylaatio ja amidihydrolyysi triatsolikarboksyylihappometaboliitin tuottamiseksi. Ribaviriini ja sen triatsolikarboksamidi ja triatsolikarboksyylihappometaboliitit erittyvät munuaisten kautta. Kun 600 mg 14C-ribaviriinia annettiin suun kautta, noin 61 % radioaktiivisuudesta poistui virtsaan ja 12 % ulosteeseen 336 tunnissa. Muuttumattoman ribaviriinin osuus oli 17 % annetusta annoksesta.
Erikoispopulaatiot
Munuaisten toimintahäiriö
Ribaviriinin farmakokinetiikka arvioitiin sen jälkeen, kun ribaviriinia annettiin kerta-annoksena suun kautta (400 mg) potilaille, joilla ei ollut HCV-infektoitunutta munuaisten vajaatoimintaa. Keskimääräinen AUCtf-arvo oli kolminkertainen koehenkilöillä, joiden kreatiniinipuhdistuma oli 10-30 ml/min, verrattuna kontrollihenkilöihin (kreatiniinipuhdistuma yli 90 ml/min). Koehenkilöillä, joiden kreatiniinipuhdistuma oli välillä 30-60 ml/min, AUCtf oli kaksinkertainen verrokkihenkilöihin verrattuna. Suurentunut AUCtf näyttää johtuvan munuaispuhdistuman ja ei-renaalisen puhdistuman vähenemisestä näillä potilailla. Vaiheen 3 tehokkuuskokeet sisälsivät koehenkilöitä, joiden kreatiniinipuhdistuma oli yli 50 ml/min. Ribaviriinin usean annoksen farmakokinetiikkaa ei voida ennustaa tarkasti potilailla, joilla on munuaisten vajaatoiminta. Ribaviriini ei poistu tehokkaasti hemodialyysillä.
Potilaita, joiden kreatiniinipuhdistuma on alle 50 ml/min, ei tule hoitaa REBETOLilla [ks. VASTA-AIHEET ].
Maksan toimintahäiriö
Maksan toimintahäiriön vaikutus arvioitiin ribaviriinin (600 mg) oraalisen kerta-annoksen jälkeen. Keskimääräiset AUCtf-arvot eivät eronneet merkittävästi koehenkilöillä, joilla oli lievä, kohtalainen tai vaikea maksan vajaatoiminta (Child-Pugh-luokitukset A, B tai C) verrokkihenkilöihin verrattuna. Keskimääräiset Cmax-arvot nousivat kuitenkin maksan toimintahäiriön vaikeusasteen myötä ja olivat kaksinkertaiset potilailla, joilla oli vaikea maksan vajaatoiminta, verrattuna kontrollihenkilöihin.
Iäkkäät potilaat
Iäkkäillä henkilöillä ei ole tehty farmakokineettisiä arviointeja.
Sukupuoli
Kliinisesti merkittäviä farmakokineettisiä eroja ei havaittu kerta-annostutkimuksessa, johon osallistui 18 miestä ja 18 naista.
Pediatriset potilaat
REBETOL 200 mg kapseleiden ja INTRON A:n usean annoksen farmakokineettiset ominaisuudet 5–16-vuotiailla lapsilla, joilla on krooninen hepatiitti C, on esitetty taulukossa 12. REBETOL 200 mg:n ja INTRON A:n (annosnormalisoitu) farmakokinetiikka on samanlainen aikuisilla lasten aiheet.
REBETOL-oraaliliuoksen täydellisiä farmakokineettisiä ominaisuuksia ei ole määritetty lapsipotilailla. Ribaviriinin Cmin-arvot olivat samanlaiset REBETOL-oraaliliuoksen tai REBETOL 200 mg -kapseleiden antamisen jälkeen 48 viikon hoidon aikana lapsipotilailla (3–16-vuotiaat).
Kliininen tutkimus 3–17-vuotiailla lapsilla, joilla oli krooninen hepatiitti C, suoritettiin, ja siinä arvioitiin PegIntronin ja REBETOLin (kapselit ja oraaliliuos) farmakokinetiikkaa. Lapsipotilailla, jotka saivat kehon pinta-alaltaan mukautettua PegIntron-annosta 60 mcg/m²/viikko, logaritmin muunnetun suhteen arvio altistumisesta annosteluvälin aikana oli 58 % [90 % CI: 141 %, 177 %] korkeampi kuin havaittiin aikuisilla, jotka saivat 1,5 mikrog/kg/viikko. REBETOLin (annosnormalisoitu) farmakokinetiikka tässä tutkimuksessa oli samanlainen kuin aiemmassa tutkimuksessa, jossa REBETOL 200 mg yhdistelmänä INTRON A:n kanssa tehtiin lapsilla ja aikuisilla.
Ruoan vaikutus ribaviriinin imeytymiseen
Sekä AUCtf että Cmax nousivat 70 %, kun REBETOL-kapseleita annettiin runsasrasvaisen aterian yhteydessä (841 kcal, 53,8 g rasvaa, 31,6 g proteiinia ja 57,4 g hiilihydraattia) kerta-annoksen farmakokineettisessä tutkimuksessa [ks. ANNOSTELU JA HALLINNOINTI ].
Mikrobiologia
Toimintamekanismi
Mekanismia, jolla ribaviriini edistää sen antiviraalista tehoa klinikalla, ei ole täysin ymmärretty. Ribaviriinilla on suora antiviraalinen vaikutus kudosviljelmässä monia RNA-viruksia vastaan. Ribaviriini lisää mutaatiotaajuutta useiden virusten genomissa ja ribaviriinitrifosfaatti estää HCV-polymeraasia biokemiallisessa reaktiossa.
Antiviraalinen aktiivisuus soluviljelmässä
Ribaviriinin antiviraalista aktiivisuutta HCV-replikonissa ei tunneta hyvin eikä sitä ole määritelty ribaviriinin solutoksisuuden vuoksi. Suoraa antiviraalista aktiivisuutta on havaittu muiden RNA-virusten kudosviljelmässä. Interferonin anti-HCV-aktiivisuus osoitettiin solussa, joka sisälsi itsestään replikoituvan HCV-RNS:n (HCV-replikonisolut) tai HCV-infektion.
Resistanssi
HCV-genotyypit osoittavat suurta vaihtelua vasteessa pegyloituun rekombinantti ihmisen interferoni/ribaviriinihoitoon. Muuttuvaan vasteeseen liittyviä geneettisiä muutoksia ei ole tunnistettu.
Ristivastus
Pegyloitujen/pegyloimattomien interferonien ja ribaviriinin välillä ei ole raportoitu ristiresistenssiä.
Eläinten toksikologia ja farmakologia
Pitkäaikaiset tutkimukset hiirellä ja rotalla [18-24 kuukautta; annokset 20 - 75 ja 10 - 40 mg/kg/vrk (arvioidut ihmisen vastaavat annokset 1,67 - 6,25 ja 1,43 - 5,71 mg/kg/vrk, 60 kg painoisen aikuisen kehon pinta-alan mukautuksen perusteella; noin 0,1-0,4 kertaa ihmisen 24 tunnin ribaviriinin enimmäisannos)] ovat osoittaneet yhteyden kroonisen ribaviriinialtistuksen ja lisääntyneiden verisuonivaurioiden (mikroskooppisten verenvuotojen) esiintyvyyden välillä hiirillä. Rotilla verkkokalvon rappeutumista esiintyi kontrolleilla, mutta esiintyvyys lisääntyi ribavirilla hoidetuilla rotilla.
Tutkimuksessa, jossa rotanpennuille annettiin postnataalisesti ribaviriinia annoksilla 10, 25 ja 50 mg/kg/vrk, huumeisiin liittyviä kuolemia esiintyi annoksella 50 mg/kg (rotan poikasten plasmapitoisuudet, jotka olivat pienempiä kuin ihmisen plasman pitoisuudet ihmisillä terapeuttinen annos) tutkimuspäivien 13 ja 48 välillä. Synnytyksen jälkeisistä päivistä 7 - 63 annetut rotanpennut osoittivat vähäistä, annoksesta riippuvaa kokonaiskasvun laskua kaikilla annoksilla, mikä ilmeni myöhemmin lievänä painon, kruunun ja lantion pituuden, ja luun pituus. Nämä vaikutukset osoittivat palautuvuutta, eikä luuhun kohdistuvia histopatologisia vaikutuksia havaittu. Ribaviriinin vaikutuksia hermostoon käyttäytymiseen tai lisääntymiseen ei havaittu.
Kliiniset tutkimukset
Kliinisessä tutkimuksessa 1 arvioitiin PegIntron-monoterapia. Katso PegIntron-merkintä saadaksesi tietoa tästä kokeesta.
REBETOL/PegIntron-yhdistelmähoito
Aikuiset aiheet
Tutkimus 2
Satunnaistetussa tutkimuksessa verrattiin hoitoa kahteen PegIntron/REBETOL 200 mg hoito-ohjelmaan [PegIntron 1,5 mikrogrammaa/kg ihonalaisesti kerran viikossa/REBETOL 800 mg suun kautta päivittäin (jaettuina annoksina); PegIntron 1,5 mcg/kg ihonalaisesti kerran viikossa 4 viikon ajan, sitten 0,5 mcg/kg ihonalaisesti kerran viikossa 44 viikon ajan/REBETOL 1000 tai 1200 mg suun kautta päivittäin (jaettuina annoksina)] INTRON A:n kanssa [3 MIU ihonalaisesti kolme kertaa viikossa1/REBETOL 1200 mg suun kautta päivässä (jaettuina annoksina)] 1530 aikuisella, joilla oli krooninen hepatiitti C. Aiemmin interferonihoitoa saamattomia koehenkilöitä hoidettiin 48 viikon ajan ja seurattiin 24 viikon ajan hoidon jälkeen. Tukikelpoisilla koehenkilöillä oli kompensoitunut maksasairaus, havaittavissa oleva HCV-RNA, kohonnut ALAT ja maksan histopatologia, joka oli yhdenmukainen kroonisen hepatiitin kanssa.
Vaste hoitoon määriteltiin havaitsemattomana HCV-RNA:na 24 viikkoa hoidon jälkeen (katso taulukko 13). PegIntron 1,5 mcg/kg ja ribaviriini 800 mg annoksen vasteprosentti oli suurempi kuin vasteprosentti INTRON A/REBETOLiin (katso taulukko 13). PegIntron 1,5 → 0,5 mcg/kg/REBETOL 200 mg:n vasteprosentti oli olennaisesti sama vastauksena INTRON A/REBETOLiin (tietoja ei näytetä).
Potilailla, joilla oli virusgenotyyppi 1, oli viruskuormasta riippumatta pienempi vaste PegIntron (1,5 mcg/kg)/REBETOL (800 mg) annokseen verrattuna henkilöihin, joilla oli muita virusgenotyyppejä. Koehenkilöillä, joilla oli sekä huonot prognostiset tekijät (genotyyppi 1 että suuri viruskuorma), vasteprosentti oli 30 % (78/256) verrattuna INTRON A/REBETOL -yhdistelmähoitoon 29 %:iin (71/247).
Koehenkilöillä, joilla oli pienempi ruumiinpaino, oli yleensä korkeampi haittavaikutusaste [katso HAITTAVAIKUTUKSET ja korkeampi vasteprosentti kuin koehenkilöillä, joilla on suurempi ruumiinpaino. Erot vastemäärissä hoitoryhmien välillä eivät vaihdelleet olennaisesti painon mukaan.
Hoitovaste PegIntron/REBETOL-yhdistelmähoidolla oli 49 % miehillä ja 56 % naisilla. Vastausprosentti oli alhaisempi afroamerikkalaisilla ja latinalaisamerikkalaisilla ja korkeampi aasialaisilla kuin valkoihoisilla. Vaikka afroamerikkalaisilla oli suurempi osuus huonoista prognostisista tekijöistä kuin valkoihoisilla, tutkittujen muiden kuin valkoihoisten määrä (11 % kokonaismäärästä) ei ollut riittävä, jotta voitaisiin tehdä mielekkäitä johtopäätöksiä vasteiden eroista sen jälkeen, kun ennustetekijöitä oli mukautettu tässä tutkimuksessa.
Maksabiopsiat otettiin ennen hoitoa ja hoidon jälkeen 68 %:lta koehenkilöistä. Lähtötasoon verrattuna noin kahdella kolmasosalla koehenkilöistä kaikissa hoitoryhmissä havaittiin lievästi vähentynyt tulehdus.
Tutkimus 3
Suuressa yhdysvaltalaistutkimuksessa 4913 kroonista hepatiitti C:tä sairastavaa henkilöä satunnaistettiin saamaan PegIntronia 1,5 mikrogrammaa/kg ihonalaisesti kerran viikossa yhdessä 800–1400 mg:n REBETOL 200 mg:n annoksen kanssa (painoon perustuva annostus [WBD]) tai 800 mg (tasainen) suun kautta päivittäin (jaettuina annoksina) 24 tai 48 viikon ajan genotyypin mukaan. Hoitovaste määriteltiin havaitsemattomana HCV-RNA:na (perustuu määritykseen, jonka havaitsemisraja oli 125 IU/ml) 24 viikkoa hoidon jälkeen.
Hoito PegIntronilla 1,5 mcg/kg ja REBETOL 800–1400 mg johti korkeampaan jatkuvaan virologiseen vasteeseen verrattuna PegIntronilla yhdistettynä tasaiseen 800 mg:n vuorokausiannokseen REBETOLia. Yli 105 kg painavat koehenkilöt saivat suurimman hyödyn WBD:stä, vaikkakin vaatimaton hyöty havaittiin myös yli 85-105 kg painavilla koehenkilöillä (katso taulukko 14). WBD:n hyöty yli 85 kg painavilla koehenkilöillä havaittiin HCV-genotyypeillä 1-3. Muita genotyyppejä koskevien päätelmien tekemiseen ei ollut riittävästi tietoa. WBD:n käyttö johti lisääntyneeseen anemian ilmaantuvuuteen [katso HAITTAVAIKUTUKSET ].
Yhteensä 1552 yli 65 kg painavalla tutkimuksessa 3 koehenkilöllä oli genotyyppi 2 tai 3, ja heidät satunnaistettiin saamaan 24 tai 48 viikon hoitoa. Lisähyötyä ei havaittu pidennetyllä hoidon kestolla.
Tutkimus 4
Laajassa satunnaistetussa tutkimuksessa verrattiin 48 viikon hoidon turvallisuutta ja tehokkuutta kahdella PegIntron/REBETOL 200 mg hoito-ohjelmalla [PegIntron 1,5 mcg/kg ja 1 mcg/kg ihonalaisesti kerran viikossa, molemmat yhdessä REBETOL 800-1400 mg:aan PO vuorokaudessa annokset)] ja Pegasys 180 mcg ihonalaisesti kerran viikossa yhdessä Copegusin kanssa 1000-1200 mg PO vuorokaudessa (kahdessa annoksessa) 3070 aiemmin hoitamattomalle aikuiselle, joilla on krooninen hepatiitti C genotyyppi 1. Tässä tutkimuksessa varhaisen virologisen vasteen puuttuminen (ei havaittavissa) HCV-RNA tai suurempi tai yhtä suuri kuin 2 log10 lasku lähtötasosta) hoidolla Viikko 12 oli hoidon lopettamisen kriteeri. SVR määriteltiin havaitsemattomaksi HCV-RNA:ksi (Roche COBAS TaqMan -määritys, kvantifioinnin alaraja 27 IU/ml) 24 viikkoa hoidon jälkeen (katso taulukko 15).
SVR:n kokonaismäärät olivat samanlaiset kolmessa hoitoryhmässä. Hoitoryhmästä riippumatta SVR-luvut olivat alhaisemmat potilailla, joilla oli huonot ennustetekijät. Koehenkilöt, joilla oli huonot prognostiset tekijät, satunnaistettiin PegIntron (1,5 mikrog/kg)/REBETOL- tai Pegasys/Copegus-hoitoon, mutta saavuttivat korkeammat SVR-luvut verrattuna vastaaviin henkilöihin, jotka satunnaistettiin saamaan PegIntron 1 mikrog/kg/REBETOL. PegIntron 1,5 mcg/kg ja REBETOL-annoksilla SVR-luvut koehenkilöillä, joilla oli ja joilla ei ollut seuraavia ennustetekijöitä, olivat seuraavat: kirroosi (10 % vs. 42 %), normaalit ALAT-tasot (32 % vs. 42 %), lähtötilanteen virustauti kuormitus yli 600 000 IU/ml (35 % vs. 61 %), 40-vuotiaat ja sitä vanhemmat (38 % vs. 50 %) ja afroamerikkalainen rotu (23 % vs. 44 %). Potilailla, joiden HCV-RNA:ta ei havaittu hoitoviikolla 12 ja jotka saivat PegIntronia (1,5 mcg/kg)/REBETOL 200 mg, SVR-aste oli 81 % (328/407).
Tutkimus 5 – REBETOL/PegIntron-yhdistelmähoito aiempien hoidon epäonnistumisissa
Ei-vertailevassa tutkimuksessa 2 293 potilasta, joilla oli kohtalainen tai vaikea fibroosi, joille aiempi alfa-interferoni/ribaviriini-yhdistelmähoito epäonnistui, hoidettiin uudelleen PegIntronilla, 1,5 mikrogrammaa/kg ihonalaisesti kerran viikossa, yhdessä painosovitetun ribaviriinin kanssa. Tukikelpoisiin koehenkilöihin kuuluivat aiemmin reagoimattomat (potilaat, jotka olivat HCV-RNA-positiivisia vähintään 12 viikon hoidon lopussa) ja aiemmin uusiutuneita (henkilöt, jotka olivat HCVRNA-negatiivisia vähintään 12 viikon hoidon lopussa ja jotka sen jälkeen uusiutuivat hoidon jälkeen seuranta). Koehenkilöitä, jotka olivat negatiivisia viikolla 12, hoidettiin 48 viikkoa ja seurattiin 24 viikkoa hoidon jälkeen. Hoitovaste määriteltiin havaitsemattomaksi HCV-RNA:ksi 24 viikkoa hoidon jälkeen (mitattu tutkimukseen perustuvalla testillä, havaitsemisraja 125 IU/ml). Kokonaisvasteprosentti oli 22 % (497/2293) (99 % CI: 19,5, 23,9). Koehenkilöt, joilla oli seuraavat ominaisuudet, hyötyivät vähemmän todennäköisesti uusintahoidosta: aiempi vaste, aikaisempi pegyloitu interferonihoito, merkittävä siltafibroosi tai kirroosi ja genotyypin 1 infektio.
Taulukossa 16 on yhteenveto uudelleenhoidon jatkuvan virologisen vasteen perusominaisuuksien mukaan.
Havaimattoman HCV-RNA:n saavuttaminen hoitoviikolla 12 oli vahva SVR:n ennustaja. Tässä tutkimuksessa 1470 (64 %) koehenkilöä ei saavuttanut havaitsematonta HCV-RNA:ta hoitoviikolla 12, ja heille tarjottiin ilmoittautumista pitkäaikaisiin hoitotutkimuksiin riittämättömän hoitovasteen vuoksi. Niistä 823 (36 %) koehenkilöstä, joiden HCV-RNA:ta ei ollut havaittavissa hoitoviikolla 12, genotyypin 1 infektoituneiden SVR oli 48 % (245/507) ja fibroosipistemäärät (F4-F2) olivat erilaisia. 39-55 %. Koehenkilöillä, joilla oli genotyyppi 2/3 infektoitunut ja joiden HCV-RNA:ta ei ollut havaittavissa hoitoviikolla 12, kokonais-SVR oli 70 % (196/281) ja vastealue fibroosipisteytyksen mukaan (F4-F2) oli 60-83 %. Kaikissa genotyypeissä korkeammat fibroosipisteet liittyivät SVR:n saavuttamisen todennäköisyyteen.
Pediatriset aiheet
Aikaisemmin hoitamattomia 3–17-vuotiaita lapsipotilaita, joilla oli kompensoitunut krooninen hepatiitti C ja havaittavissa oleva HCV-RNA, hoidettiin REBETOLilla 15 mg/kg/vrk ja PegIntronilla 60 mcg/m² kerran viikossa 24 tai 48 viikon ajan HCV:n genotyypin ja lähtötilanteen viruksen perusteella. ladata. Kaikkia koehenkilöitä seurattiin 24 viikon ajan hoidon jälkeen. Yhteensä 107 koehenkilöä sai hoitoa, joista 52 % oli naisia, 89 % valkoihoisia ja 67 % oli infektoituneita HCV:n genotyypillä 1. Genotyypeillä 1, 4 tai genotyypeillä 3 infektoituneet koehenkilöt, joiden HCV-RNA oli suurempi tai yhtä suuri kuin 600 000 IU/ml saivat 48 viikon hoitoa, kun taas ne, joilla oli genotyyppi 2 tai genotyyppi 3, joiden HCV-RNA:ta oli alle 600 000 IU/ml, saivat 24 viikon hoitoa. Koetulokset on koottu taulukkoon 17.
REBETOL/INTRON A yhdistelmähoito
Aikuiset aiheet
Aikaisemmin hoitamattomat kohteet
Aikuiset, joilla oli kompensoitu krooninen hepatiitti C ja havaittavissa oleva HCV-RNA (arvioitu keskuslaboratoriossa käyttämällä tutkimukseen perustuvaa RT-PCR-määritystä), joita ei aiemmin ollut hoidettu alfainterferonihoidolla, otettiin mukaan kahteen monikeskustutkimukseen (USA ja kansainvälinen) ja satunnaistettiin saamaan REBETOL 200 mg kapseleita 1200 mg/vrk (1000 mg/vrk henkilöille, jotka painavat enintään 75 kg) ja INTRON A 3 MIU kolmesti viikossa tai INTRON A ja lumelääke 24 tai 48 viikon ajan, jota seuraa 24 viikkoa terapian ulkopuolinen seuranta. Kansainvälinen tutkimus ei sisältänyt 24-viikkoista INTRON A:ta ja lumelääkettä. Yhdysvaltalaiseen tutkimukseen osallistui 912 henkilöä, joista 67 % oli lähtötilanteessa miehiä, 89 % valkoihoisia ja keskimääräinen Knodell HAI -pistemäärä (I+II+III) oli 7,5 ja 72 % genotyyppi 1. Kansainvälinen tutkimus suoritettiin Euroopassa, Israelissa. Kanadassa ja Australiassa otettiin mukaan 799 koehenkilöä (65 % miehiä, 95 % valkoihoisia, keskimääräinen Knodell-pistemäärä 6,8 ja 58 % genotyyppi 1).
Koetuloksista on yhteenveto taulukossa 18.
Koehenkilöistä, jotka eivät olleet saavuttaneet HCV-RNA:ta alle tutkimukseen perustuvan määrityksen havaintorajan REBETOL/INTRON A -hoidon viikkoon 24 mennessä, alle 5 % reagoi 24 viikon yhdistelmähoitoon.
REBETOL/INTRON A -hoidolla hoidetuista HCV-genotyypin 1 koehenkilöistä, jotka saavuttivat HCV-RNA:n alle tutkimukseen perustuvan määrityksen havaitsemisrajan 24 viikkoon mennessä, 48 viikon hoitoon satunnaistettujen potilaiden virologiset vasteet olivat korkeammat kuin 24-vuotiailla. viikon hoitoryhmä. Vastausasteen nousua ei havaittu potilailla, joilla oli HCV ei-genotyyppi 1 ja jotka satunnaistettiin REBETOL/INTRON A -hoitoon 48 viikon ajan verrattuna 24 viikkoon.
Relapse aiheet
Koehenkilöt, joilla oli kompensoitu krooninen hepatiitti C ja havaittavissa oleva HCV-RNA (arvioitu keskuslaboratoriossa tutkimukseen perustuvalla RT-PCR-määrityksellä), jotka olivat uusiutuneet yhden tai kahden interferonihoitojakson jälkeen (määritelty epänormaaleiksi seerumin ALT-tasoiksi), otettiin mukaan kahteen ryhmään. monikeskustutkimukset, kaksoissokkotutkimukset (Yhdysvallat ja kansainväliset) ja satunnaistettiin saamaan REBETOL 1200 mg/vrk (1000 mg/vrk henkilöille, jotka painavat ≤ 75 kg) ja INTRON A 3 MIU kolmesti viikossa tai INTRON A ja lumelääke 24 viikon ajan, minkä jälkeen 24 viikkoa hoidon ulkopuolista seurantaa. Yhdysvaltalaiseen tutkimukseen osallistui 153 henkilöä, joista 67 % oli lähtötilanteessa miehiä, 92 % valkoihoisia ja keskimääräinen Knodell HAI -pistemäärä (I+II+III) oli 6,8 ja 58 % genotyyppi 1. Kansainvälinen tutkimus suoritettiin Euroopassa, Israelissa. Kanadassa ja Australiassa otettiin mukaan 192 koehenkilöä (64 % miehiä, 95 % valkoihoisia, keskimääräinen Knodell-pistemäärä 6,6 ja 56 % genotyyppi 1). Koetuloksista on yhteenveto taulukossa 19.
Virologiset ja histologiset vasteet olivat samanlaisia miehillä ja naisilla sekä aiemmin hoitamattomissa että uusiutuneissa tutkimuksissa.
Pediatriset aiheet
3–16-vuotiaita lapsipotilaita, joilla oli kompensoitunut krooninen hepatiitti C ja havaittavissa oleva HCV-RNA (arvioitu keskuslaboratorio käyttäen tutkimukseen perustuvaa RT-PCR-määritystä), hoidettiin REBETOLilla 15 mg/kg päivässä ja INTRON A:lla 3 MIU/ m² kolme kertaa viikossa 48 viikon ajan, jota seuraa 24 viikon hoidon ulkopuolinen seuranta. Hoitoa sai yhteensä 118 henkilöä, joista 57 % oli miehiä, 80 % valkoihoisia ja 78 % genotyyppi 1. Alle 5-vuotiaat saivat REBETOL-oraaliliuosta ja 5-vuotiaat tai sitä vanhemmat joko REBETOL-oraaliliuosta tai kapselit.
Koetulokset on koottu taulukkoon 20.
Koehenkilöillä, joilla oli virusgenotyyppi 1, viruskuormasta riippumatta, oli pienempi vaste INTRON A/REBETOL -yhdistelmähoitoon verrattuna koehenkilöihin, joiden genotyyppi oli non-1, 36 % vs. 81 %. Koehenkilöillä, joilla oli molemmat huonot prognostiset tekijät (genotyyppi 1 ja korkea viruskuorma), vasteprosentti oli 26 % (13/50).
POTILASTIEDOT
Tietoja ei ole annettu. Katso VAROITUKSET JA VAROTOIMET osio.