Epivir 150mg Lamivudine Käyttö, sivuvaikutukset ja annostus. Hinta verkkoapteekissa. Geneeriset lääkkeet ilman reseptiä.
Mitä Epivir on ja miten sitä käytetään?
Epivir 150 mg on reseptilääke, jota käytetään HIV-infektion ja kroonisen hepatiitti B:n oireiden hoitoon. Epivir 150 mg voidaan käyttää yksinään tai muiden lääkkeiden kanssa.
Epivir kuuluu lääkeluokkaan, jota kutsutaan hepatiitti B:n/hepatiitti C:n aiheuttajiksi. HIV, NRTI:t.
Ei tiedetä, onko Epivir turvallinen ja tehokas alle 3 kuukauden ikäisille lapsille.
Mitkä ovat Epivir 150 mg:n mahdolliset sivuvaikutukset?
Epivir voi aiheuttaa vakavia sivuvaikutuksia, mukaan lukien:
- nokkosihottuma,
- vaikeuksia hengittää,
- kasvojen, huulten, kielen tai kurkun turvotus,
- epätavallinen lihaskipu,
- vatsakipu,
- oksentelua,
- epäsäännöllinen syke,
- huimaus,
- kylmä olo,
- heikkous,
- väsymys,
- voimakas kipu ylävatsassa, joka leviää selkään,
- pahoinvointi,
- nopea syke,
- turvotus keskiosan ympärillä,
- oikeanpuoleinen ylävatsakipu,
- ruokahalun menetys,
- tumma virtsa,
- savenväriset ulosteet,
- ihon tai silmien keltaisuus (keltatauti),
- kuume,
- yöhikoilut,
- turvonneet rauhaset,
- huuliherpes,
- yskä,
- hengityksen vinkuminen,
- ripuli,
- painonpudotus,
- puhe- tai nielemisvaikeuksia,
- ongelmia tasapainossa tai silmien liikkeessä,
- piikikäs tunne,
- turvotus niskassa tai kurkussa (kilpirauhasen suureneminen),
- kuukautiskierron muutokset ja
- impotenssi
Hakeudu välittömästi lääkärin hoitoon, jos sinulla on jokin yllä mainituista oireista.
Epivirin yleisimpiä sivuvaikutuksia ovat:
- pahoinvointi,
- ripuli,
- päänsärky,
- kuume,
- väsymys,
- yleinen huono olo,
- korvakipu tai täyteläinen tunne,
- kuulohäiriöt,
- vuoto korvasta,
- lapsen kiukkuisuus,
- tukkoinen nenä,
- aivastelu,
- kurkkukipu ja
- yskä
Kerro lääkärille, jos sinulla on haittavaikutuksia, jotka häiritsevät sinua tai jotka eivät häviä.
Nämä eivät ole kaikkia Epivirin mahdollisia sivuvaikutuksia. Lisätietoja saat lääkäriltäsi tai apteekista.
Soita lääkärillesi saadaksesi lääketieteellisiä neuvoja sivuvaikutuksista. Voit ilmoittaa sivuvaikutuksista FDA:lle numerossa 1-800-FDA-1088.
VAROITUS
Hepatiitti B:n paheneminen ja EPIVIRIN ERI FORMAALIT.
Hepatiitti B:n pahenemisvaiheet
Hepatiitti B:n vakavia akuutteja pahenemisvaiheita on raportoitu potilailla, joilla on samanaikaisesti hepatiitti B-virus (HBV) ja ihmisen immuunikatovirus (HIV-1) ja jotka ovat lopettaneet EPIVIR-hoidon. Maksan toimintaa on seurattava tarkasti sekä kliinisen että laboratoriotutkimuksen avulla vähintään useiden kuukausien ajan potilailla, jotka lopettavat EPIVIR-hoidon ja joilla on samanaikaisesti HIV-1- ja HBV-infektio. Tarvittaessa hepatiitti B:n vastaisen hoidon aloittaminen voi olla perusteltua [katso VAROITUKSET JA VAROTOIMET].
Tärkeitä eroja lamivudiinia sisältävien tuotteiden välillä
EPIVIR 150 mg tabletit ja oraaliliuos (käytetään HIV-1-infektion hoitoon) sisältävät suuremman annoksen vaikuttavaa ainetta (lamivudiinia) kuin EPIVIR-HBV-tabletit ja oraaliliuos (käytetään kroonisen HBV-infektion hoitoon). Potilaiden, joilla on HIV-1-infektio, tulisi saada vain HIV-1:n hoitoon sopivia annosmuotoja (ks. VAROITUKSET JA VAROTOIMET).
KUVAUS
EPIVIR (tunnetaan myös nimellä 3TC) on tuotenimi lamivudiinille, synteettiselle nukleosidianalogille, jolla on vaikutusta HIV-1:tä ja HBV:tä vastaan. Lamivudiinin kemiallinen nimi on (2R,cis)-4-amino-1(2-hydroksimetyyli-1,3-oksatiolan-5-yyli)-(1 H)-pyrimidin-2-oni. Lamivudiini on sytidiinin dideoksianalogin (-)enantiomeeri. Lamivudiinia on kutsuttu myös (-)2',3'-dideoksiksi, 3'tiasytidiiniksi. Sen molekyylikaava on C8H11N3O3S ja molekyylipaino 229,3 g per mooli. Sillä on seuraava rakennekaava:
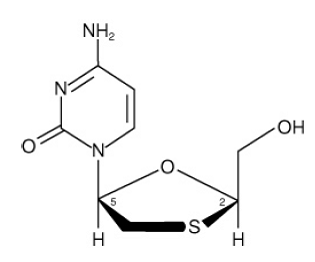
Lamivudiini on valkoinen tai luonnonvalkoinen kiteinen kiinteä aine, jonka liukoisuus on noin 70 mg/ml veteen 20 °C:ssa.
EPIVIR 150 mg tabletit ovat suun kautta otettavia. Jokainen uurrettu 150 mg:n kalvopäällysteinen tabletti sisältää 150 mg lamivudiinia ja inaktiivisia aineita hypromelloosia, magnesiumstearaattia, mikrokiteistä selluloosaa, polyetyleeniglykolia, polysorbaatti 80:tä, natriumtärkkelysglykolaattia ja titaanidioksidia.
Jokainen 300 mg:n kalvopäällysteinen tabletti sisältää 300 mg lamivudiinia ja inaktiivisia aineosia mustaa rautaoksidia, hypromelloosia, magnesiumstearaattia, mikrokiteistä selluloosaa, polyetyleeniglykolia, polysorbaatti 80:tä, natriumtärkkelysglykolaattia ja titaanidioksidia.
EPIVIR-oraaliliuos on tarkoitettu annettavaksi suun kautta. Yksi millilitra (1 ml) EPIVIR-oraaliliuosta sisältää 10 mg lamivudiinia (10 mg/ml) vesiliuoksessa ja inaktiivisia aineosia keinotekoisia mansikka- ja banaaniaromeja, sitruunahappoa (vedetön), metyyliparabeenia, propyleeniglykolia, propyyliparabeenia, natriumsitraattia. (dihydraatti) ja sakkaroosi (200 mg).
INDIKAATIOT
EPIVIR 150mg on nukleosidianalogi, joka on tarkoitettu yhdistelmänä muiden antiretroviraalisten aineiden kanssa ihmisen immuunikatoviruksen tyypin 1 (HIV-1) infektion hoitoon.
Käyttörajoitukset
- Tämän tuotteen annostus on tarkoitettu HIV-1:lle eikä HBV:lle.
ANNOSTELU JA HALLINNOINTI
Suositeltu annostus aikuisille potilaille
Suositeltu EPIVIR 150 mg:n annos HIV-1-tartunnan saaneille aikuisille on 300 mg päivässä, joko 150 mg:na suun kautta kahdesti vuorokaudessa tai 300 mg:na suun kautta kerran vuorokaudessa ruoan kanssa tai ilman. Jos lamivudiinia annetaan HIV-1- ja HBV-tartunnan saaneelle potilaalle, HIV-1-hoitoon osoitettua annosta tulee käyttää osana sopivaa yhdistelmähoitoa [ks. VAROITUKSET JA VAROTOIMET ].
Suositeltu annostus lapsipotilaille
EPIVIR 150 mg:n jakouurteinen tabletti on suositeltu lääkemuoto HIV-1-tartunnan saaneille lapsipotilaille, jotka painavat vähintään 14 kg ja joille sopiva annosmuoto on sopiva. Ennen uurrettujen EPIVIR-tablettien määräämistä lapsipotilaiden kyky niellä tabletteja on arvioitava. Potilaille, jotka eivät pysty nielemään EPIVIR 150 mg tabletteja turvallisesti ja luotettavasti, voidaan määrätä oraaliliuosmuoto [ks. VAROITUKSET JA VAROTOIMET ]. Suositeltu suun kautta otettava EPIVIR 150 mg tablettien annos HIV-1-tartunnan saaneille lapsipotilaille on esitetty taulukossa 1.
Oraaliliuos
Suositeltu EPIVIR 150 mg oraaliliuoksen annostus HIV-1-tartunnan saaneille vähintään 3 kuukauden ikäisille lapsipotilaille on 5 mg/kg suun kautta kahdesti vuorokaudessa tai 10 mg/kg suun kautta kerran vuorokaudessa (enintään 300 mg päivässä), annetaan yhdessä muiden antiretroviraalisten aineiden kanssa [katso KLIININEN FARMAKOLOGIA ]. Harkitse HIV-1-viruskuormaa ja CD4+-solujen määrää/prosenttia, kun valitset annosväliä potilaille, jotka aloittavat hoidon oraaliliuoksella [ks. VAROITUKSET JA VAROTOIMET , KLIININEN FARMAKOLOGIA ].
Potilaat, joilla on munuaisten vajaatoiminta
EPIVIR 150 mg:n annostus säädetään munuaisten toiminnan mukaan. Annoksen säädöt on lueteltu taulukossa 2 [katso KLIININEN FARMAKOLOGIA ].
EPIVIR 150 mg:n lisäannostusta ei tarvita rutiininomaisen (4 tunnin) hemodialyysin tai peritoneaalidialyysin jälkeen.
Vaikka tiedot eivät ole riittäviä, jotta voitaisiin suositella erityistä EPIVIR-annoksen muuttamista munuaisten vajaatoimintaa sairastaville lapsipotilaille, annoksen pienentämistä ja/tai annosteluvälin pidentämistä tulee harkita.
MITEN TOIMITETAAN
Annostusmuodot ja vahvuudet
EPIVIR-tabletit
EPIVIR-uurteiset tabletit sisältävät 150 mg lamivudiinia. Tabletit ovat valkoisia, vinoneliön muotoisia, jakouurteisia, kalvopäällysteisiä tabletteja, joiden molemmilla puolilla on merkintä "GX CJ7".
EPIVIR-tabletit
EPIVIR-tabletit sisältävät 300 mg lamivudiinia. Tabletit ovat harmaita, muunneltuja timantin muotoisia, kalvopäällysteisiä, ja niiden toiselle puolelle on kaiverrettu "GX EJ7" ja toiselle puolelle sileä.
EPIVIR oraaliliuos
EPIVIR oraaliliuos sisältää 10 mg lamivudiinia 1 ml:ssa. Liuos on kirkas, väritön tai vaaleankeltainen, mansikan ja banaanin makuinen neste.
Varastointi ja käsittely
EPIVIR jakouurteiset tabletit sisältävät 150 mg lamivudiinia, ovat valkoisia, vinoneliön muotoisia, kalvopäällysteisiä, ja niissä on molemmilla puolilla merkintä ”GX CJ7”. Pakattu seuraavasti:
Pullossa 60 tablettia ( NDC 49702-203-18) lapsiturvallisella sulkimella.
EPIVIR Tabletit sisältävät 300 mg lamivudiinia, ovat harmaita, muokatun vinoneliön muotoisia, kalvopäällysteisiä, ja niiden toisella puolella on kaiverrus "GX EJ7" ja toisella puolella sileä. Pakattu seuraavasti:
Pullossa 30 tablettia ( NDC 49702-204-13) lapsiturvallisella sulkimella.
Suositeltu säilytystila
Säilytä EPIVIR-tabletit 25 °C:ssa (77 °F); retket sallitaan 15° - 30°C (59° - 86°F) [katso USP-ohjattu huonelämpötila ].
EPIVIR oraaliliuos on kirkas, väritön tai vaaleankeltainen, mansikan ja banaanin makuinen neste. Jokainen ml liuosta sisältää 10 mg lamivudiinia. Pakattu seuraavasti:
Muovipullo 240 ml (NDC 49702-205-48), jossa lapsiturvallinen suljin. Tämä tuote ei vaadi liuottamista.
Säilytä tiiviisti suljetuissa pulloissa 25 °C:ssa (77 °F) [katso USP-ohjattu huonelämpötila ].
Valmistettu: ViiV Healthcare Research Triangle Park, NC 27709. tekijä: GlaxoSmithKline Research Triangle Park, NC 27709, Valmistettu Shire Pharmaceuticals Group plc:n sopimuksella. Tarkistettu: huhtikuuta 2018
SIVUVAIKUTUKSET
Seuraavia haittavaikutuksia käsitellään muissa merkintöjen osissa:
- Hepatiitti B:n paheneminen [katso LAATIKKO VAROITUS , VAROITUKSET JA VAROTOIMET ].
- Maitohappoasidoosi ja vaikea hepatomegalia, johon liittyy steatoosi [katso VAROITUKSET JA VAROTOIMET ].
- Maksan vajaatoiminta potilailla, joilla on samanaikaisesti HIV-1- ja hepatiitti C -infektio [katso VAROITUKSET JA VAROTOIMET ].
- Haimatulehdus [katso VAROITUKSET JA VAROTOIMET ].
- Immuunirekonstituutio-oireyhtymä [katso VAROITUKSET JA VAROTOIMET ].
Kokemus kliinisistä kokeista
Kokemus kliinisistä kokeista aikuisilla
Koska kliiniset tutkimukset suoritetaan hyvin vaihtelevissa olosuhteissa, lääkkeen kliinisissä tutkimuksissa havaittuja haittavaikutuksia ei voida suoraan verrata toisen lääkkeen kliinisissä tutkimuksissa havaittuihin nopeuksiin, eivätkä ne välttämättä kuvasta kliinisessä käytännössä havaittuja nopeuksia.
EPIVIR 150 mg:n turvallisuusprofiili aikuisilla perustuu ensisijaisesti 3 568 HIV-1-tartunnan saaneeseen henkilöön 7 kliinisessä tutkimuksessa.
Yleisimmät haittavaikutukset ovat päänsärky, pahoinvointi, huonovointisuus, väsymys, nenän merkit ja oireet, ripuli ja yskä.
Taulukossa 3 on lueteltu valikoidut kliiniset haittavaikutukset, jotka esiintyivät vähintään 5 %:lla potilaista EPIVIR 150 mg kahdesti vuorokaudessa ja 200 mg RETROVIR 3 kertaa vuorokaudessa hoidon aikana 24 viikon ajan.
Haimatulehdus
Haimatulehdus havaittiin yhdeksällä 2 613 aikuisesta (0,3 %), jotka saivat EPIVIR 150 mg kontrolloiduissa kliinisissä tutkimuksissa EPV20001, NUCA3001, NUCB3001, NUCA3002, NUCB3002 ja NUCB3007 [ks. VAROITUKSET JA VAROTOIMET ].
EPIVIR 300 mg kerran päivässä
Kliinisten haittavaikutusten tyypit ja esiintymistiheydet olivat samankaltaisia 48 viikon ajan EPIVIR 300 mg kerran vuorokaudessa tai EPIVIR 150 mg kahdesti vuorokaudessa saaneilla (kolmen lääkkeen yhdistelmähoitona EPV20001:ssä ja EPV40001:ssä).
Taulukossa 4 on yhteenveto valituista hoidon aikana havaituista laboratoriopoikkeavuuksista.
Valittujen laboratoriotulosten poikkeavuuksien esiintymistiheydet olivat samankaltaisia potilailla, jotka saivat EPIVIR 300 mg kerran vuorokaudessa tai EPIVIR 150 mg kahdesti vuorokaudessa (kolmen lääkkeen yhdistelmähoidoissa EPV20001:ssä ja EPV40001:ssä).
Kokemus kliinisistä kokeista lapsipotilailla
EPIVIR-oraaliliuosta on tutkittu 638:lla 3 kuukauden–18-vuotiaalla lapsipotilaalla kolmessa kliinisessä tutkimuksessa.
Valitut kliiniset haittavaikutukset ja fyysiset löydökset, joiden esiintymistiheys on suurempi tai yhtä suuri kuin 5 % hoidon aikana EPIVIR-annoksella 4 mg/kg kahdesti vuorokaudessa plus RETROVIR 160 mg/m² 3 kertaa vuorokaudessa, kun hoitoa ei ole käytetty (vähemmän kuin 56 päivää antiretroviraalista hoitoa) terapia) lapsipotilaat on lueteltu taulukossa 5.
Haimatulehdus
Haimatulehdus, joka on joissakin tapauksissa johtanut kuolemaan, on havaittu lapsilla, jotka ovat aiemmin saaneet antiretroviraalista nukleosideja, jotka ovat saaneet EPIVIRiä yksinään tai yhdessä muiden antiretroviraalisten aineiden kanssa. Avoimessa annoskorotustutkimuksessa (NUCA2002) 14 koehenkilölle (14 %) kehittyi haimatulehdus EPIVIR-monoterapian aikana. Kolme näistä koehenkilöistä kuoli haimatulehduksen komplikaatioihin. Toisessa avoimessa tutkimuksessa (NUCA2005) 12 koehenkilölle (18 %) kehittyi haimatulehdus. ACTG300-tutkimuksessa haimatulehdusta ei havaittu 236 potilaalla, jotka satunnaistettiin saamaan EPIVIR 150 mg plus RETROVIR. Haimatulehdus havaittiin tässä tutkimuksessa yhdellä koehenkilöllä, joka sai avoimen EPIVIRin yhdessä RETROVIRin ja ritonaviirin kanssa didanosiinimonoterapian lopettamisen jälkeen [ks. VAROITUKSET JA VAROTOIMET ].
Parestesiat ja perifeeriset neuropatiat
Parestesioita ja perifeerisiä neuropatioita raportoitiin 15 koehenkilöllä (15 %) tutkimuksessa NUCA2002, 6 koehenkilöllä (9 %) tutkimuksessa NUCA2005 ja 2 koehenkilöllä (alle 1 %) tutkimuksessa ACTG300.
Taulukossa 6 on lueteltu valikoidut laboratorioarvojen poikkeavuudet, joita kokevat aiemmin hoitoa saamattomat (alle tai yhtä suuri kuin 56 päivää antiretroviraalista hoitoa) lapsilla.
Pediatriset koehenkilöt kerran päivässä vs. kahdesti päivässä annostelu (COL105677)
ARROW-tutkimuksessa arvioitiin kerran vuorokaudessa antamisen turvallisuutta verrattuna 150 mg:n EPIVIR:n kahdesti vuorokaudessa antoon. Ensisijainen turvallisuusarviointi ARROW-tutkimuksessa perustui asteen 3 ja asteen 4 haittatapahtumiin. Asteiden 3 ja 4 haittavaikutusten esiintymistiheys oli samanlainen potilailla, jotka satunnaistettiin saamaan kerran vuorokaudessa annettavaa lääkettä, verrattuna kahdesti vuorokaudessa saaneisiin. Yhden asteen 4 hepatiittitapahtuman kerran päivässä saaneessa kohortissa tutkija piti epävarmana syy-yhteyteen, ja tutkija katsoi, että kaikki muut asteen 3 tai 4 haittatapahtumat eivät liittyneet toisiinsa.
Vastasyntyneet
Rajoitetut lyhytaikaiset turvallisuustiedot ovat saatavilla kahdesta pienestä, kontrolloimattomasta Etelä-Afrikassa tehdystä tutkimuksesta vastasyntyneillä, jotka saivat lamivudiinia tsidovudiinin kanssa tai ilman sitä ensimmäisen elinviikon ajan äidin hoidon jälkeen alkaen raskausviikolla 38 tai 36 [ks. KLIININEN FARMAKOLOGIA ]. Näillä vastasyntyneillä raportoituja tiettyjä haittavaikutuksia olivat kohonneet maksan toimintakokeet, anemia, ripuli, elektrolyyttihäiriöt, hypoglykemia, keltaisuus ja hepatomegalia, ihottuma, hengitystieinfektiot ja sepsis; 3 vastasyntynyttä kuoli (1 gastroenteriittiin, johon liittyi asidoosi ja kouristuksia, 1 traumaattisesta vammasta ja 1 tuntemattomista syistä). Kaksi muuta ei-fataalista gastroenteriitti- tai ripulitapausta raportoitiin, mukaan lukien yksi kouristuksia; Yhdellä lapsella oli kuivumiseen liittyvä ohimenevä munuaisten vajaatoiminta. Kontrolliryhmien puuttuminen rajoittaa syy-yhteyden arviointia, mutta on oletettava, että perinataalisesti altistuneilla imeväisillä saattaa olla riski saada haittavaikutuksia, jotka ovat verrattavissa lamivudiinia sisältävillä yhdistelmähoidoilla hoidetuilla lapsilla ja aikuisilla HIV-1-infektiopotilailla raportoituihin haittavaikutuksiin. In utero ja lapsen lamivudiinialtistuksen pitkäaikaisvaikutuksia ei tunneta.
Markkinoinnin jälkeinen kokemus
Seuraavat haittavaikutukset on tunnistettu EPIVIRin hyväksymisen jälkeisen käytön aikana. Koska nämä reaktiot on raportoitu vapaaehtoisesti tuntemattoman kokoiselta populaatiolta, ei ole aina mahdollista luotettavasti arvioida niiden esiintymistiheyttä tai määrittää syy-yhteyttä lääkealtistukseen. Nämä reaktiot on valittu sisällytettäviksi niiden vakavuuden, ilmoitustiheyden tai mahdollisen syy-yhteyden lamivudiiniin yhdistelmän vuoksi.
Keho kokonaisuutena
Kehon rasvan uudelleenjakautuminen/kertyminen.
Endokriininen ja aineenvaihdunta
Hyperglykemia.
Kenraali
Heikkous. Heminen ja lymfaattinen anemia (mukaan lukien puhdas punasoluaplasia ja vaikeat anemiat, jotka etenevät hoidon aikana).
Maksa ja haima
Maitohappoasidoosi ja maksan steatoosi [katso VAROITUKSET JA VAROTOIMET ], hepatiitti B:n hoidon jälkeiset pahenemisvaiheet [katso VAROITUKSET JA VAROTOIMET ].
Yliherkkyys
Anafylaksia, urtikaria.
Tuki- ja liikuntaelimistön
Lihasheikkous, CPK:n nousu, rabdomyolyysi. Ihon hiustenlähtö, kutina.
HUUMEIDEN VUOROVAIKUTUKSET
Orgaanisia kationinkuljettajia estävät lääkkeet
Lamivudiini eliminoituu pääasiassa virtsaan aktiivisen orgaanisen kationisen erityksen kautta. Yhteisvaikutusten mahdollisuus muiden samanaikaisesti annettavien lääkkeiden kanssa tulee ottaa huomioon, erityisesti kun niiden pääasiallinen eliminaatioreitti on aktiivinen munuaiseritys orgaanisen kationisen kuljetusjärjestelmän kautta (esim. trimetopriimi) [ks. KLIININEN FARMAKOLOGIA ]. Tietoja yhteisvaikutuksista muiden lääkkeiden kanssa, joiden munuaispuhdistuma on samanlainen kuin lamivudiinilla, ei ole saatavilla.
sorbitoli
Lamivudiinin ja sorbitolin kerta-annosten samanaikainen anto johti sorbitoliannoksesta riippuvaiseen lamivudiinialtistuksen pienenemiseen. Vältä sorbitolia sisältävien lääkkeiden käyttöä lamivudiinin kanssa mahdollisuuksien mukaan [katso VAROITUKSET JA VAROTOIMET , KLIININEN FARMAKOLOGIA ].
VAROITUKSET
Mukana osana VAROTOIMENPITEET osio.
VAROTOIMENPITEET
Potilaat, joilla on samanaikainen hepatiitti B -virusinfektio
Hepatiitin pahenemisvaiheet hoidon jälkeen
Kliinisiä ja laboratoriotutkimuksia hepatiitin pahenemisesta on havaittu lamivudiinihoidon lopettamisen jälkeen. Nämä pahenemisvaiheet on havaittu ensisijaisesti seerumin ALAT-arvojen nousuna HBV-DNA:n uudelleen ilmaantumisen lisäksi. Vaikka useimmat tapahtumat näyttävät rajoittuneen itsestään, joissakin tapauksissa on raportoitu kuolemantapauksia. Samanlaisia tapahtumia on raportoitu markkinoille tulon jälkeen, kun lamivudiinia sisältävistä HIV-1-hoito-ohjelmista on vaihdettu ei-lamivudiinia sisältäviä hoito-ohjelmia potilailla, joilla on sekä HIV-1- että HBV-infektio. Syy-yhteyttä lamivudiinihoidon lopettamiseen ei tunneta. Potilaita tulee seurata tarkasti sekä kliinisen että laboratoriotutkimuksen avulla vähintään useiden kuukausien ajan hoidon lopettamisen jälkeen.
Tärkeitä eroja lamivudiinia sisältävien tuotteiden välillä
EPIVIR 150 mg tabletit ja oraaliliuos sisältävät suuremman annoksen samaa vaikuttavaa ainetta (lamivudiinia) kuin EPIVIR-HBV tabletit ja EPIVIR-HBV oraaliliuos. EPIVIR-HBV kehitettiin potilaille, joilla on krooninen hepatiitti B. Lamivudiinin formulaatio ja annostus EPIVIR-HBV:ssä ei sovellu potilaille, joilla on samanaikaisesti HIV-1- ja HBV-infektio. Lamivudiinin turvallisuutta ja tehoa ei ole osoitettu kroonisen hepatiitti B:n hoidossa potilailla, joilla on samanaikaisesti HIV-1- ja HBV-infektio. Jos EPIVIR-HBV-hoitoa määrätään krooniseen B-hepatiittiin potilaalle, jolla on tunnistamaton tai hoitamaton HIV-1-infektio, seurauksena on todennäköisesti nopea HIV-1-resistenssin ilmaantuminen subterapeuttisen annoksen ja HIV-1-monoterapiahoidon sopimattomuuden vuoksi. Jos päätetään antaa lamivudiinia potilaille, joilla on samanaikaisesti HIV-1- ja HBV-infektio, EPIVIR 150 mg tabletteja, EPIVIR 150 mg oraaliliuosta tai muuta valmistetta, joka sisältää suuremman annoksen lamivudiinia, tulee käyttää osana sopivaa yhdistelmähoitoa.
Lamivudiiniresistentin HBV:n ilmaantuminen
Lamivudiinin turvallisuutta ja tehoa ei ole varmistettu kroonisen hepatiitti B:n hoidossa potilailla, joilla on kaksoisinfektio HIV-1- ja HBV-infektiolla (katso EPIVIR-HBV:n täydelliset reseptitiedot). Lamivudiiniresistenssiin liittyvien hepatiitti B -viruksen varianttien ilmaantumista on raportoitu myös HIV-1-infektoituneilla henkilöillä, jotka ovat saaneet lamivudiinia sisältäviä antiretroviraalisia hoitoja, kun hepatiitti B -virusinfektio on samanaikainen.
Maitohappoasidoosi ja vaikea hepatomegalia ja steatoosi
Maitohappoasidoosia ja vaikeaa hepatomegaliaa, johon liittyy steatoosia, mukaan lukien kuolemaan johtaneet tapaukset, on raportoitu käytettäessä nukleosidianalogeja, mukaan lukien EPIVIR. Suurin osa näistä tapauksista on ollut naisilla. Naisten sukupuoli ja liikalihavuus voivat olla riskitekijöitä maitohappoasidoosin ja vaikean hepatomegalian kehittymiselle, johon liittyy steatoosia antiretroviraalisilla nukleosidianalogeilla hoidetuilla potilailla. EPIVIR-hoito tulee keskeyttää jokaiselle potilaalle, jolle kehittyy kliiniset tai laboratoriolöydökset, jotka viittaavat maitohappoasidoosiin tai voimakkaaseen maksatoksisuuteen, joihin voi sisältyä hepatomegalia ja steatoosi, vaikka transaminaasiarvot eivät kohoaisi merkittävästi.
Käytä interferoni- ja ribaviriinipohjaisten hoito-ohjelmien kanssa
In vitro -tutkimukset ovat osoittaneet, että ribaviriini voi vähentää pyrimidiininukleosidianalogien, kuten lamivudiinin, fosforylaatiota. Vaikka mitään näyttöä farmakokineettisistä tai farmakodynaamisista yhteisvaikutuksista (esim. HIV-1/HCV-virologisen suppression häviämisestä) ei havaittu, kun ribaviriinia annettiin samanaikaisesti lamivudiinin kanssa HIV-1/HCV-yhteisinfektion saaneilla potilailla [ks. KLIININEN FARMAKOLOGIA ], maksan vajaatoimintaa (jossain määrin kuolemaan johtanutta) on esiintynyt HIV-1/HCV-yhteisinfektion saaneilla potilailla, jotka ovat saaneet HIV-1:n ja interferoni alfan yhdistelmähoitoa ribaviriinin kanssa tai ilman. Potilaita, jotka saavat interferoni alfaa ribaviriinin ja EPIVIRin kanssa tai ilman niitä, tulee seurata tarkasti hoitoon liittyvien toksisten vaikutusten, erityisesti maksan vajaatoiminnan, varalta. EPIVIR 150 mg -hoidon lopettamista tulee harkita lääketieteellisesti tarkoituksenmukaisena. Interferoni alfan, ribaviriinin tai molempien annoksen pienentämistä tai lopettamista tulee myös harkita, jos havaitaan kliinisen toksisuuden pahenemista, mukaan lukien maksan vajaatoiminta (esim. Child-Pugh yli 6). Katso kaikki interferonin ja ribaviriinin reseptitiedot.
Haimatulehdus
Lapsipotilailla, joilla on aiemmin ollut antiretroviraalisille nukleosidille altistuminen, haimatulehdus tai muita merkittäviä haimatulehduksen kehittymisen riskitekijöitä, EPIVIR 150 mg:aa tulee käyttää varoen. EPIVIR 150mg -hoito tulee lopettaa välittömästi, jos ilmenee haimatulehdukseen viittaavia kliinisiä merkkejä, oireita tai laboratorioarvojen poikkeavuuksia [ks. HAITTAVAIKUTUKSET ].
Immuunijärjestelmän palautumisoireyhtymä
Immuunirekonstituution oireyhtymää on raportoitu potilailla, joita on hoidettu antiretroviraalisella yhdistelmähoidolla, mukaan lukien EPIVIR. Antiretroviraalisen yhdistelmähoidon alkuvaiheessa potilaat, joiden immuunijärjestelmä reagoi, voivat kehittää tulehdusreaktion indolenteille tai jäljellä oleville opportunistisille infektioille (kuten Mycobacterium avium -infektio, sytomegalovirus, Pneumocystis jirovecii -keuhkokuume [PCP] tai tuberkuloosi), mikä saattaa edellyttää lisäarviointia. ja hoitoon.
Autoimmuunisairauksia (kuten Gravesin tauti, polymyosiitti ja Guillain-Barrén oireyhtymä) on myös raportoitu esiintyvän immuunijärjestelmän palautumisen yhteydessä, mutta aika niiden alkamiseen on vaihtelevampi ja voi ilmaantua useita kuukausia hoidon aloittamisen jälkeen.
Alempi virologinen suppressio ja lisääntynyt virusresistenssin riski oraaliliuoksella
Lapsipotilailla, jotka saivat EPIVIR 150 mg oraaliliuosta (painoaluekohtaisina annoksina, noin 8 mg/kg/vrk) samanaikaisesti muiden antiretroviraalisten oraaliliuosten kanssa milloin tahansa ARROW-tutkimuksessa, oli alhaisempi virologinen suppressio, pienempi plasman lamivudiinialtistus, ja he kehittyivät virusresistenssi useammin kuin EPIVIR-tabletteja saaneet [ks KLIININEN FARMAKOLOGIA , Mikrobiologia , Kliiniset tutkimukset ].
EPIVIR-uurrettu tabletti on suositeltu lääkemuoto HIV-1-tartunnan saaneille lapsipotilaille, jotka painavat vähintään 14 kg ja joille sopiva annosmuoto on sopiva. Kaikkia tabletteja sisältävää hoito-ohjelmaa tulee käyttää mahdollisuuksien mukaan mahdollisen yhteisvaikutuksen välttämiseksi sorbitolin kanssa [katso KLIININEN FARMAKOLOGIA ]. Harkitse HIV-1-viruskuorman tiheämpää seurantaa, kun hoidetaan EPIVIR 150 mg oraaliliuoksella.
Potilasneuvontatiedot
Neuvo potilasta lukemaan FDA:n hyväksymä potilasmerkintä (potilastiedot).
Potilaat, joilla on samanaikainen hepatiitti B- tai C-infektio
Kerro potilaille, joilla on samanaikaisesti HIV-1- ja HBV-infektio, että maksasairaus on pahentunut joissakin tapauksissa, kun lamivudiinihoito lopetettiin. Neuvo potilaita keskustelemaan kaikista hoito-ohjelman muutoksista terveydenhuollon tarjoajan kanssa [katso VAROITUKSET JA VAROTOIMET ].
Kerro potilaille, joilla on samanaikainen HIV-1/HCV-infektio, että maksan vajaatoimintaa (jossain määrin kuolemaan johtavaa) on ilmennyt HIV-1/HCV-yhteisinfektiopotilailla, jotka saavat HIV-1:n ja interferoni alfan yhdistelmähoitoa ribaviriinin kanssa tai ilman sitä [ks. VAROITUKSET JA VAROTOIMET ].
EPIVIR-formulaatioiden erot
Kerro potilaille, että EPIVIR 150 mg tabletit ja oraaliliuos sisältävät suuremman annoksen samaa vaikuttavaa ainetta (lamivudiinia) kuin EPIVIR-HBV-tabletit ja oraaliliuos. Jos päätetään sisällyttää lamivudiinia HIV-1-hoitoon potilaalla, jolla on samanaikaisesti HIV-1- ja HBV-infektio, on käytettävä EPIVIR:ssä (ei EPIVIR-HBV:ssä) olevan lamivudiinin formulaatiota ja annosta [ks. VAROITUKSET JA VAROTOIMET ].
Maitohappoasidoosi/hepatomegalia ja steatoosi
Kerro potilaille, että maitohappoasidoosia ja vaikeaa hepatomegaliaa, johon liittyy steatoosia, on raportoitu käytettäessä nukleosidianalogeja ja muita antiretroviraalisia lääkkeitä. Neuvo potilaita lopettamaan EPIVIRin käyttö, jos heille kehittyy kliinisiä oireita, jotka viittaavat maitohappoasidoosiin tai voimakkaaseen maksatoksisuuteen [ks. VAROITUKSET JA VAROTOIMET ].
Haimatulehduksen riski
Neuvo vanhempia tai huoltajia seuraamaan lapsipotilaita haimatulehduksen merkkien ja oireiden varalta [katso VAROITUKSET JA VAROTOIMET ].
Immuunijärjestelmän palautumisoireyhtymä
Neuvo potilaita ilmoittamaan terveydenhuollon tarjoajalleen välittömästi kaikista infektion merkeistä ja oireista, koska aikaisemman infektion aiheuttama tulehdus voi ilmaantua pian antiretroviraalisen yhdistelmähoidon jälkeen, mukaan lukien EPIVIR-hoidon aloittaminen [ks. VAROITUKSET JA VAROTOIMET ].
Alempi virologinen suppressio ja lisääntynyt virusresistenssin riski oraaliliuoksella
Neuvo potilaita, että kaikkien tablettien hoito-ohjelmaa tulee käyttää mahdollisuuksien mukaan, koska hoidon epäonnistuminen lisääntyi lapsilla, jotka saivat EPIVIR-oraaliliuosta samanaikaisesti muiden antiretroviraalisten oraaliliuosten kanssa [ks. VAROITUKSET JA VAROTOIMET ].
EPIVIR-oraaliliuoksen sakkaroosipitoisuus
Kerro diabeetikoille, että jokainen 15 ml:n annos EPIVIR-oraaliliuosta sisältää 3 grammaa sakkaroosia (1 ml = 200 mg sakkaroosia) [ks. KUVAUS ].
Raskausrekisteri
Kerro potilaille, että on olemassa raskausaltistusrekisteri, joka seuraa raskauden tuloksia naisilla, jotka ovat altistuneet EPIVIRille raskauden aikana [ks. Käyttö tietyissä populaatioissa ].
Imetys
Neuvo naisia, joilla on HIV-1-infektio, olemaan imettämättä, koska HIV-1 voi siirtyä vauvaan äidinmaidon mukana [ks. Käyttö tietyissä populaatioissa ].
Unohtunut annos
Neuvo potilaita ottamaan se heti kun muistavat, jos he unohtavat ottaa EPIVIR 150 mg -annoksen. Neuvo potilaita olemaan kaksinkertaistamatta seuraavaa annosta tai ottamasta enemmän kuin määrätty annosta [katso ANNOSTELU JA HALLINNOINTI ].
EPIVIR ja RETROVIR ovat ViiV Healthcare -konsernin omistamia tai niille lisensoituja tavaramerkkejä.
Ei-kliininen toksikologia
Karsinogeneesi, mutageneesi, hedelmällisyyden heikkeneminen
Karsinogeneesi
Pitkäaikaiset karsinogeenisuustutkimukset lamivudiinilla hiirillä ja rotilla eivät osoittaneet karsinogeenisuuspotentiaalia altistuksilla, jotka olivat korkeintaan 10-kertaisia (hiiret) ja 58-kertaisia (rotat) verrattuna ihmisen altistukseen suositellulla 300 mg:n annoksella.
Mutageneesi
Lamivudiini oli mutageeninen L5178Y-hiiren lymfoomamäärityksessä ja klastogeeninen sytogeneettisessä määrityksessä, jossa käytettiin viljeltyjä ihmisen lymfosyyttejä. Lamivudiini ei ollut mutageeninen mikrobien mutageenisuuskokeessa, in vitro solutransformaatiomäärityksessä, rotan mikrotumatestissä, rotan luuytimen sytogeneettisessä määrityksessä eikä määrityksessä, jossa määritettiin suunnittelematon DNA-synteesi rotan maksassa. Lamivudiini ei osoittanut näyttöä in vivo genotoksisesta aktiivisuudesta rotilla oraalisilla annoksilla, jotka olivat korkeintaan 2 000 mg/kg, jolloin plasmapitoisuudet olivat 35-45 kertaa suuremmat kuin ihmisillä suositellulla HIV-1-infektion annoksella.
Hedelmällisyyden heikkeneminen
Lisääntymiskykyä koskevassa tutkimuksessa lamivudiinia annettiin rotille annoksina 4 000 mg/kg/vrk, jolloin plasmapitoisuudet olivat 47-70 kertaa suuremmat kuin ihmisillä, ei osoittanut merkkejä hedelmällisyyden heikkenemisestä eikä vaikutusta eloonjäämiseen, kasvuun ja kehitykseen. jälkeläisten vieroittamiseen.
Käyttö tietyissä populaatioissa
Raskaus
Raskausaltistusrekisteri
On olemassa raskausaltistusrekisteri, joka seuraa raskauden tuloksia naisilla, jotka ovat altistuneet EPIVIRille raskauden aikana. Terveydenhuollon tarjoajia rohkaistaan rekisteröimään potilaita soittamalla Antiretroviral Pregnancy Registry (APR) numeroon 1-800-258-4263.
Riskien yhteenveto
Saatavilla olevat APR-tiedot eivät osoita eroa lamivudiinin synnynnäisten epämuodostumien yleisessä riskissä verrattuna synnynnäisten epämuodostumien taustatasoon, joka on 2,7 % Metropolitan Atlanta Congenital Defects -ohjelman (MACDP) vertailupopulaatiossa (katso Data ). Todellinen vuosiluku käyttää MACDP:tä Yhdysvaltain vertailupopulaationa yleisen väestön synnynnäisten epämuodostumien osalta. MACDP arvioi naisia ja imeväisiä rajoitetulta maantieteelliseltä alueelta, eikä se sisällä tuloksia alle 20 raskausviikolla tapahtuneista synnytyksistä. Keskenmenojen määrää ei ilmoiteta vuosikorkoon. Arvioitu keskenmenon taustaaste kliinisesti tunnistetuissa raskauksissa Yhdysvaltojen väestössä on 15–20 %. Taustariskiä vakaville synnynnäisille epämuodostumille ja keskenmenolle ilmoitetulle väestölle ei tunneta.
Eläinten lisääntymistutkimuksissa lamivudiinin oraalinen antaminen tiineille kaniineille organogeneesin aikana johti alkiokuolleisuuteen systeemisellä altistuksella (AUC), joka oli samanlainen kuin suositeltu kliininen annos. haitallisia kehitysvaikutuksia ei kuitenkaan havaittu annettaessa lamivudiinia suun kautta tiineille rotille organogeneesin aikana plasmapitoisuuksilla (Cmax) 35 kertaa suositeltu kliininen annos (ks. Data ).
Data
Ihmistiedot
APR:ään perustuvien ennustettujen raporttien mukaan yli 11 000 lamivudiinialtistusta raskauden aikana, mikä johti elävänä syntymiseen (mukaan lukien yli 4 500 ensimmäisen raskauskolmanneksen aikana altistunutta), lamivudiinin synnynnäisten epämuodostumien kokonaisriskin ja taustasyntyneiden epämuodostumien välillä ei ollut eroa. 2,7 % Yhdysvaltain MACDP:n vertailupopulaatiossa. Defektien esiintyvyys elävänä syntyneissä oli 3,1 % (95 % CI: 2,6 % - 3,6 %) ensimmäisen raskauskolmanneksen altistuksen jälkeen lamivudiinia sisältäville hoito-ohjelmille ja 2,8 % (95 % CI: 2,5 % - 3,3 %) toisen/kolmannen raskauskolmanneksen altistuksen jälkeen lamivudiinia sisältäviin hoito-ohjelmiin.
Lamivudiinin farmakokinetiikkaa tutkittiin raskaana olevilla naisilla kahdessa Etelä-Afrikassa tehdyssä kliinisessä tutkimuksessa. Tutkimuksissa arvioitiin farmakokinetiikka 16 naisella 36 raskausviikolla käyttämällä 150 mg lamivudiinia kahdesti vuorokaudessa tsidovudiinin kanssa, 10 naisella 38 raskausviikolla 150 mg lamivudiinia kahdesti vuorokaudessa tsidovudiinin kanssa ja 10 naisella 38 raskausviikolla kahdesti 300 mg lamivudiinia. päivittäin ilman muita antiretroviruslääkkeitä. Näitä tutkimuksia ei ole suunniteltu tai käytetty antamaan tehotietoja. Lamivudiinipitoisuudet olivat yleensä samanlaiset äidin, vastasyntyneen ja napanuoran seeruminäytteissä. Osasta koehenkilöistä otettiin lapsivesinäytteitä kalvojen luonnollisen repeämisen jälkeen, ja ne vahvistivat, että lamivudiini läpäisee istukan ihmisillä. Rajallisten synnytystietojen perusteella lamivudiinin lapsivesipitoisuuden mediaani (vaihteluväli) oli 3,9 (1,2-12,8)-kertainen verrattuna äidin seerumin paripitoisuuteen (n = 8).
Eläinten tiedot
Lamivudiinia annettiin suun kautta raskaana oleville rotille (90, 600 ja 4 000 mg/kg/vrk) ja kaneille (90, 300 ja 1000 mg/kg/vrk ja 15, 40 ja 90 mg/kg/vrk) organogeneesin aikana (raskauspäivinä 7-16 [rotta] ja 8-20 [kani]). Lamivudiinista johtuvia sikiön epämuodostumia ei havaittu rotilla ja kaniineilla annoksilla, jotka tuottivat plasman pitoisuudet (Cmax) noin 35 kertaa suuremmat kuin ihmisen altistus suositellulla vuorokausiannoksella. Todisteita varhaisesta alkiokuolleisuudesta havaittiin kaniineilla systeemisillä altistuksilla (AUC), jotka olivat samankaltaisia kuin ihmisillä, mutta tästä vaikutuksesta ei ollut viitteitä rotilla plasmapitoisuuksilla (Cmax), jotka olivat 35 kertaa suuremmat kuin ihmisen altistus suositellulla vuorokausiannoksella. . Tiineillä rotilla tehdyt tutkimukset osoittivat, että lamivudiini siirtyy sikiöön istukan kautta. Rotilla tehdyssä hedelmällisyyden/pre- ja postnataalisen kehityksen tutkimuksessa lamivudiinia annettiin suun kautta annoksina 180, 900 ja 4 000 mg/kg/vrk (ennen parittelua 20. postnataaliseen päivään). Tutkimuksessa lamivudiinin antaminen äidille ei vaikuttanut jälkeläisten kehitykseen, hedelmällisyyteen ja lisääntymiskykyyn mukaan lukien.
Imetys
Riskien yhteenveto
Centers for Disease Control and Prevention suosittelee, että Yhdysvalloissa HIV-1-tartunnan saaneet äidit eivät imetä lapsiaan välttääkseen HIV-1-infektion postnataalisen leviämisen riskin. Lamivudiini erittyy äidinmaitoon. Lamivudiinin vaikutuksista imetettävään lapseen tai lääkkeiden vaikutuksista maidontuotantoon ei ole tietoa. Koska (1) HIV-1-tartunta (HIV-negatiivisilla imeväisillä), (2) virusresistenssin kehittyminen (HIV-positiivisilla imeväisillä) ja (3) rintaruokitetulla lapsella voi esiintyä haittavaikutuksia, neuvo äitejä olemaan imettämättä. jos he saavat EPIVIRiä.
Käyttö lapsille
EPIVIR 150 mg:n turvallisuus ja tehokkuus yhdessä muiden antiretroviraalisten aineiden kanssa on osoitettu 3 kuukauden ikäisillä ja sitä vanhemmilla lapsipotilailla. EPIVIR-uurrettu tabletti on suositeltava formulaatio HIV-1-tartunnan saaneille lapsipotilaille, jotka painavat vähintään 14 kg ja joille sopiva annosmuoto on sopiva, koska lapsipotilailla, jotka saivat EPIVIR 150 mg oraaliliuosta, virologinen suppressio ja plasman lamivudiinialtistus olivat alhaisemmat. ja kehittyivät virusresistenssiä useammin kuin EPIVIR 150 mg tabletteja saaneilla ARROW-tutkimuksessa [ks. ANNOSTELU JA HALLINNOINTI , VAROITUKSET JA VAROTOIMET , HAITTAVAIKUTUKSET , KLIININEN FARMAKOLOGIA , Kliiniset tutkimukset ].
Geriatrinen käyttö
EPIVIRin kliinisissä tutkimuksissa ei ollut riittävästi 65-vuotiaita ja sitä vanhempia koehenkilöitä sen määrittämiseksi, reagoivatko he eri tavalla kuin nuoremmat. Yleensä varovaisuutta tulee noudattaa EPIVIR-valmisteen antamisessa iäkkäille potilaille, koska maksan, munuaisten tai sydämen toiminta on heikentynyt sekä samanaikainen sairaus tai muu lääkehoito [ks. ANNOSTELU JA HALLINNOINTI , KLIININEN FARMAKOLOGIA ].
Potilaat, joilla on munuaisten vajaatoiminta
EPIVIR 150 mg:n annoksen pienentämistä suositellaan potilaille, joilla on munuaisten vajaatoiminta [ks. ANNOSTELU JA HALLINNOINTI , KLIININEN FARMAKOLOGIA ].
YLIANNOSTUS
EPIVIRin yliannostukseen ei tunneta erityistä hoitoa. Jos yliannostus tapahtuu, potilasta on seurattava ja tavanomaista tukihoitoa on sovellettava tarvittaessa. Koska vähäinen määrä lamivudiinia poistettiin (4 tunnin) hemodialyysin, jatkuvan ambulatorisen peritoneaalidialyysin ja automatisoidun peritoneaalidialyysin kautta, ei tiedetä, olisiko jatkuva hemodialyysi kliinistä hyötyä lamivudiinin yliannostustapahtumassa.
VASTA-AIHEET
EPIVIR 150 mg on vasta-aiheinen potilaille, joilla on aiemmin ollut yliherkkyysreaktio lamivudiinille.
KLIININEN FARMAKOLOGIA
Toimintamekanismi
Lamivudiini on antiretroviraalinen aine [katso Mikrobiologia ].
Farmakokinetiikka
Farmakokinetiikka aikuisilla
Lamivudiinin farmakokineettisiä ominaisuuksia on tutkittu oireettomilla, HIV-1-infektoituneilla aikuisilla koehenkilöillä 0,25–8 mg/kg:n kerta-annosten (iv) sekä kerta- ja toistuvien (kahdesti vuorokaudessa annettujen) oraalisten annosten jälkeen. välillä 0,25-10 mg/kg.
Lamivudiinin farmakokineettisiä ominaisuuksia on myös tutkittu kerta- ja toistuvina suun kautta 5 mg:sta 600 mg:aan vuorokaudessa annettuna HBV-tartunnan saaneille henkilöille.
EPIVIR 300 mg tabletin vakaan tilan farmakokineettiset ominaisuudet kerran vuorokaudessa 7 päivän ajan verrattuna EPIVIR 150 mg tablettiin kahdesti vuorokaudessa 7 päivän ajan arvioitiin ristikkäistutkimuksessa, johon osallistui 60 tervettä henkilöä. EPIVIR 300 mg kerran vuorokaudessa johti lamivudiinialtistukseen, joka oli samanlainen kuin EPIVIR 150 mg kahdesti vuorokaudessa plasman AUC24,ss:n suhteen; Cmax,ss oli kuitenkin 66 % korkeampi ja minimiarvo 53 % pienempi verrattuna 150 mg:n kahdesti vuorokaudessa annettuun hoitoon. Solunsisäiset lamivudiinitrifosfaattialtistukset perifeerisen veren mononukleaarisoluissa olivat myös samankaltaisia AUC24,ss:n ja Cmax24,ss:n suhteen; alimmaiset arvot olivat kuitenkin alhaisemmat verrattuna 150 mg kahdesti vuorokaudessa annettuun hoitoon. Koehenkilöiden välinen vaihtelu oli suurempi solunsisäisten lamivudiinitrifosfaattipitoisuuksien kohdalla verrattuna lamivudiinin plasman pienimpiin pitoisuuksiin.

Lamivudiinin farmakokinetiikkaa arvioitiin 12 aikuisella HIV-1-infektoituneella henkilöllä, joille annettiin lamivudiinia 150 mg kahdesti vuorokaudessa yhdessä muiden antiretroviraalisten aineiden kanssa. Geometrinen keskiarvo (95 % CI) AUC(0-12):lle oli 5,53 (4,58, 6,67) mcg.h/ml ja Cmax:lle 1,40 (1,17, 1,69) mcg/ml.
Imeytyminen ja biologinen hyötyosuus
Absoluuttinen hyötyosuus 12 aikuisella koehenkilöllä oli 86 % ± 16 % (keskiarvo ± SD) 150 mg:n tabletilla ja 87 % ± 13 % oraaliliuoksella. Kun 9 HIV-1-potilaalle annettiin suun kautta 2 mg/kg kahdesti päivässä, seerumin lamivudiinin huippupitoisuus (Cmax) oli 1,5 ± 0,5 mikrog/ml (keskiarvo ± SD). Plasman pitoisuus-aikakäyrän alla oleva pinta-ala (AUC) ja Cmax kasvoivat suhteessa oraaliseen annokseen alueella 0,25–10 mg/kg.
Lamivudiinin kumulaatiosuhde HIV-1-positiivisilla oireettomilla aikuisilla, joilla on normaali munuaisten toiminta, oli 1,50, kun 15 päivää annettiin suun kautta 2 mg/kg kahdesti vuorokaudessa.
Ruoan vaikutukset suun kautta imeytymiseen
EPIVIR 150 mg tabletit ja oraaliliuos voidaan ottaa ruoan kanssa tai ilman. Lamivudiinin 25 mg:n tutkimusannosmuoto annettiin suun kautta 12 oireettolle, HIV-1-tartunnan saaneelle henkilölle 2 kertaa, kerran paastotilassa ja kerran ruoan kanssa (1 099 kcal; 75 g rasvaa, 34 g proteiinia, 72 g hiilihydraattia ). Lamivudiinin imeytyminen oli hitaampaa ruokailun jälkeen (Tmax: 3,2 ± 1,3 tuntia) verrattuna tyhjään tilaan (Tmax: 0,9 ± 0,3 tuntia); Cmax oli ruokailutilassa 40 % ± 23 % (keskiarvo ± SD) pienempi kuin paastotilassa. Systeemisessä altistumisessa (AUC∞) ei ollut merkitsevää eroa ruokailun ja paaston aikana.
Jakelu
Näennäinen jakautumistilavuus lamivudiinin laskimonsisäisen annon jälkeen 20 koehenkilölle oli 1,3 ± 0,4 l/kg, mikä viittaa siihen, että lamivudiini jakautuu suonen ulkopuolisiin tiloihin. Jakaantumistilavuus oli annoksesta riippumaton eikä korreloi ruumiinpainon kanssa.
Lamivudiinin sitoutuminen ihmisen plasman proteiineihin on alle 36 %. In vitro -tutkimukset osoittivat, että pitoisuusalueella 0,1-100 mikrogrammaa/ml punasoluihin liittyvä lamivudiinin määrä vaihteli 53-57 % ja se oli pitoisuudesta riippumaton.
Aineenvaihdunta
Lamivudiinin metabolia on vähäinen eliminaatioreitti. Ihmisillä ainoa lamivudiinin tunnettu metaboliitti on trans-sulfoksidimetaboliitti (noin 5 % oraalisesta annoksesta 12 tunnin kuluttua). Tämän metaboliitin pitoisuutta seerumissa ei ole määritetty. Lamivudiini ei metaboloidu merkittävästi sytokromi P450 -entsyymien vaikutuksesta.
Eliminointi
Suurin osa lamivudiinista eliminoituu muuttumattomana virtsaan aktiivisen orgaanisen kationisen erityksen kautta. Yhdeksällä terveellä koehenkilöllä, joille annettiin kerta-annos 300 mg lamivudiinia suun kautta, munuaispuhdistuma oli 199,7 ± 56,9 ml minuutissa (keskiarvo ± SD). 20 HIV-1-tartunnan saaneella henkilöllä, joille annettiin kerta-annos laskimoon, munuaispuhdistuma oli 280,4 ± 75,2 ml/min (keskiarvo ± SD), mikä edustaa 71 % ± 16 % (keskiarvo ± SD) lamivudiinin kokonaispuhdistumasta.
Useimmissa kerta-annostutkimuksissa HIV-1-tartunnan saaneilla, HBV-tartunnan saaneilla henkilöillä tai terveillä koehenkilöillä, joista otettiin seeruminäytteitä 24 tunnin ajan annostelun jälkeen, havaittu keskimääräinen eliminaation puoliintumisaika (t½) oli 5-7 tuntia. HIV-1-tartunnan saaneilla koehenkilöillä kokonaispuhdistuma oli 398,5 ± 69,1 ml minuutissa (keskiarvo ± SD). Suun kautta otettu puhdistuma ja eliminaation puoliintumisaika olivat annoksesta ja ruumiinpainosta riippumattomia suun kautta otetulla annosalueella 0,25–10 mg/kg.
Tietyt populaatiot
Potilaat, joilla on munuaisten vajaatoiminta
Lamivudiinin farmakokineettiset ominaisuudet on määritetty pienellä ryhmällä HIV-1-tartunnan saaneita aikuisia, joilla on munuaisten vajaatoiminta (taulukko 7).
Munuaisten toiminta ei vaikuttanut merkittävästi Tmax-arvoon. Näiden havaintojen perusteella on suositeltavaa, että lamivudiinin annosta muutetaan potilailla, joilla on munuaisten vajaatoiminta [ks. ANNOSTELU JA HALLINNOINTI ].
Muutoin terveillä koehenkilöillä, joilla on heikentynyt munuaisten toiminta, tehdyn tutkimuksen perusteella hemodialyysi lisäsi lamivudiinin puhdistumaa keskimäärin 64 ml:sta 88 ml:aan minuutissa; Hemodialyysin kesto (4 tuntia) ei kuitenkaan ollut riittävä muuttamaan merkittävästi keskimääräistä lamivudiinialtistusta kerta-annoksen jälkeen. Jatkuvalla ambulatorisella peritoneaalidialyysillä ja automatisoidulla peritoneaalidialyysillä on mitätön vaikutus lamivudiinin puhdistumaan. Sen vuoksi on suositeltavaa, ettei annosta muuteta rutiininomaisen hemodialyysin tai peritoneaalidialyysin jälkeen sen jälkeen, kun kreatiniinipuhdistuma on korjattu.
Munuaisten vajaatoiminnan vaikutuksia lamivudiinin farmakokinetiikkaan lapsipotilailla ei tunneta.
Potilaat, joilla on maksan vajaatoiminta
Lamivudiinin farmakokineettiset ominaisuudet on määritetty aikuisilla, joilla on maksan vajaatoiminta. Maksan toiminnan heikkeneminen ei muuttanut farmakokineettisiä parametreja. Lamivudiinin turvallisuutta ja tehoa ei ole osoitettu dekompensoituneen maksasairauden yhteydessä.
Raskaana olevat naiset
Lamivudiinin farmakokinetiikkaa tutkittiin 36 raskaana olevalla naisella kahdessa Etelä-Afrikassa tehdyssä kliinisessä tutkimuksessa. Lamivudiinin farmakokinetiikka raskaana olevilla naisilla oli samanlainen kuin ei-raskaana olevilla aikuisilla ja synnytyksen jälkeen. Lamivudiinipitoisuudet olivat yleensä samanlaiset äidin, vastasyntyneen ja napanuoran seeruminäytteissä.
Pediatriset potilaat
Lamivudiinin farmakokinetiikkaa on tutkittu joko kerta- tai toistuvien EPIVIR-annosten jälkeen 210 lapsella. Lapsipotilaat, jotka saivat lamivudiinioraaliliuosta (annostus noin 8 mg/kg/vrk), saavuttivat noin 25 % pienemmät plasman lamivudiinipitoisuudet HIV-1-infektoituneisiin aikuisiin verrattuna. Lapsipotilaat, jotka saivat lamivudiinitabletteja suun kautta, saavuttivat plasmapitoisuudet, jotka olivat verrattavissa aikuisilla havaittuihin tai hieman korkeammat. Sekä EPIVIR-tablettien että oraaliliuoksen absoluuttinen hyötyosuus on pienempi lapsilla kuin aikuisilla. Lapsipotilailla EPIVIR-oraaliliuoksen suhteellinen hyötyosuus on noin 40 % pienempi kuin lamivudiinia sisältävien tablettien, vaikka aikuisilla ei ole eroa. Pienempi lamivudiinialtistus lapsipotilailla, jotka saavat EPIVIR 150 mg oraaliliuosta, johtuu todennäköisesti lamivudiinin ja samanaikaisten sorbitolia sisältävien liuosten (kuten ZIAGEN) välisestä yhteisvaikutuksesta. Farmakokineettisten tietojen mallinnus viittaa siihen, että EPIVIR-oraaliliuoksen annostusta on nostettava 5 mg:aan/kg suun kautta kahdesti vuorokaudessa tai 10 mg:aan/kg suun kautta kerran vuorokaudessa (enintään 300 mg/vrk), jotta saavutetaan riittävät lamivudiinipitoisuudet [ks. ANNOSTELU JA HALLINNOINTI ]. Ei ole kliinisiä tietoja HIV-1-tartunnan saaneista lapsipotilaista, jotka on annettu samanaikaisesti sorbitolia sisältävien lääkkeiden kanssa tällä annoksella.
Kerran vuorokaudessa annostellun lamivudiinin farmakokinetiikkaa HIV-1-tartunnan saaneilla lapsilla, joiden ikä oli 3 kuukauden ja 12 vuoden ikäinen, arvioitiin kolmessa tutkimuksessa (PENTA-15 [n = 17], PENTA 13 [n = 19] ja ARROW PK [n = 35]). Kaikki kolme tutkimusta olivat 2-jaksoisia, ristikkäisiä, avoimia farmakokineettisiä tutkimuksia, joissa abakaviiria ja lamivudiinia annettiin kahdesti vs. kerran päivässä. Nämä kolme tutkimusta osoittivat, että kerran vuorokaudessa annettaessa saadaan samanlainen AUC0-24 kuin kahdesti vuorokaudessa annettaessa lamivudiinia samalla kokonaisvuorokausiannoksella, kun verrataan annostusohjelmia saman formulaation (eli joko oraaliliuoksen tai tabletin) sisällä. Keskimääräinen Cmax oli noin 80–90 % korkeampi, kun lamivudiinia annettiin kerran vuorokaudessa kuin kahdesti vuorokaudessa.
Lamivudiinin jakautumista aivo-selkäydinnesteeseen (CSF) arvioitiin 38 lapsipotilaalla lamivudiinin toistuvan oraalisen annostelun jälkeen. Aivo-selkäydinnestenäytteet kerättiin 2-4 tuntia annoksen ottamisen jälkeen. Annoksella 8 mg/kg/vrk aivoselkäydinnesteen lamivudiinipitoisuudet 8 koehenkilöllä vaihtelivat välillä 5,6 % - 30,9 % (keskiarvo ± SD 14,2 % ± 7,9 %) samanaikaisen seeruminäytteen pitoisuudesta, kun aivo-selkäydinnesteen lamivudiinipitoisuudet vaihtelivat 0,04 - 0,3 mikrogrammaa per ml.
Lamivudiinin (ja tsidovudiinin) antamisesta 36:lle alle viikon ikäiselle lapselle kahdessa Etelä-Afrikassa tehdyssä tutkimuksessa on saatavilla rajoitetusti, kontrolloimatonta farmakokineettistä ja turvallisuutta koskevia tietoja. Näissä tutkimuksissa lamivudiinin puhdistuma pieneni merkittävästi 1 viikon ikäisillä vastasyntyneillä verrattuna aiemmin tutkittuihin lapsiin (yli 3 kuukauden ikäisiin). Ei ole riittävästi tietoa puhdistuman muutosten aikarajan määrittämiseksi välittömän vastasyntyneen ajanjakson ja yli 3 kuukauden ikäisten ikäryhmien välillä [ks. HAITTAVAIKUTUKSET ].
Geriatriset potilaat
Lamivudiinin farmakokinetiikkaa EPIVIR 150 mg:n antamisen jälkeen yli 65-vuotiaille henkilöille ei ole tutkittu [ks. Käyttö tietyissä populaatioissa ].
Mies- ja naispotilaat
Lamivudiinin farmakokinetiikassa ei ole merkittäviä tai kliinisesti merkittäviä sukupuolten välisiä eroja.
Roturyhmät
Lamivudiinin farmakokinetiikassa ei ole merkittäviä tai kliinisesti merkittäviä rodullisia eroja.
Huumeiden vuorovaikutustutkimukset
Lamivudiinin vaikutus muiden aineiden farmakokinetiikkaan
In vitro -tutkimustulosten perusteella lamivudiinin ei odoteta vaikuttavan terapeuttisilla lääkeainealtistuksilla sellaisten lääkkeiden farmakokinetiikkaan, jotka ovat seuraavien kuljettajien substraatteja: orgaaninen anionikuljetuspolypeptidi 1B1/3 (OATP1B1/3), rintasyöpäresistenssiproteiini (BCRP), P-glykoproteiini (P-gp), monilääke- ja toksiiniekstruusioproteiini 1 (MATE)1, MATE2-K, orgaaninen kationinkuljettaja 1 (OCT)1, OCT2 tai OCT3.
Muiden aineiden vaikutus lamivudiinin farmakokinetiikkaan
Lamivudiini on MATE1:n, MATE2-K:n ja OCT2:n substraatti in vitro. Trimetopriimin (näiden lääkekuljettajien estäjä) on osoitettu lisäävän plasman lamivudiinipitoisuuksia. Tätä yhteisvaikutusta ei pidetä kliinisesti merkittävänä, koska lamivudiiniannosta ei tarvitse muuttaa.
Lamivudiini on P-gp:n ja BCRP:n substraatti; sen absoluuttisen biologisen hyötyosuuden (87 %) huomioon ottaen on kuitenkin epätodennäköistä, että näillä kuljettajilla olisi merkittävä rooli lamivudiinin imeytymisessä. Siksi näiden effluksikuljettajien estävien lääkkeiden samanaikainen käyttö ei todennäköisesti vaikuta lamivudiinin jakautumiseen ja eliminaatioon.
Interferoni Alfa
Lamivudiinin ja interferoni alfan välillä ei havaittu merkittävää farmakokineettistä yhteisvaikutusta 19 terveen mieshenkilön tutkimuksessa [ks. VAROITUKSET JA VAROTOIMET ].
Ribaviriini
In vitro -tiedot osoittavat, että ribaviriini vähentää lamivudiinin, stavudiinin ja tsidovudiinin fosforylaatiota. Farmakokineettisiä (esim. plasmapitoisuudet tai solunsisäiset trifosforyloitujen aktiivisten metaboliittien pitoisuudet) tai farmakodynaamisia (esim. HIV-1/HCV virologisen suppression häviäminen) yhteisvaikutuksia ei havaittu, kun ribaviriini ja lamivudiini (n = 18), stavudiini (n = 10) tai tsidovudiinia (n = 6) annettiin samanaikaisesti osana usean lääkkeen hoitoa HIV-1/HCV-infektoituneille potilaille [ks. VAROITUKSET JA VAROTOIMET ].
Sorbitoli (apuaine)
Lamivudiini- ja sorbitoliliuoksia annettiin samanaikaisesti 16 terveelle aikuiselle henkilölle avoimessa, satunnaistetussa sekvenssissä, 4-jaksoisessa risteyttävässä tutkimuksessa. Jokainen koehenkilö sai yhden 300 mg:n annoksen lamivudiinioraaliliuosta yksinään tai yhdessä 3,2 gramman, 10,2 gramman tai 13,4 gramman sorbitoliannoksen kanssa liuoksessa. Lamivudiinin samanaikainen anto sorbitolin kanssa johti annoksesta riippuvaan AUC(0-24) pienenemiseen 20 %, 39 % ja 44 %, AUC(∞) 14 %, 32 % ja 36 % ja 28 %. 52 % ja 55 % Cmax:ssa; lamivudiinista.
Trimetopriimi/sulfametoksatsoli
Lamivudiinia ja TMP/SMX:ää annettiin samanaikaisesti 14 HIV-1-positiiviselle henkilölle yhden keskuksen, avoimessa, satunnaistetussa risteyttävässä tutkimuksessa. Kukin koehenkilö sai hoitoa yhdellä 300 mg:n annoksella lamivudiinia ja TMP:tä 160 mg/SMX 800 mg kerran vuorokaudessa 5 päivän ajan, ja samanaikaisesti annettiin 300 mg lamivudiinia viidennen annoksen kanssa ristikkäismallissa. TMP/SMX:n samanaikainen antaminen lamivudiinin kanssa johti lamivudiinin AUC∞:n nousuun 43 % ± 23 % (keskiarvo ± SD), lamivudiinin oraalisen puhdistuman pienenemiseen 29 % ± 13 % ja 30 % ± 36 % laskuun lamivudiinin munuaispuhdistuma. TMP:n ja SMX:n farmakokineettiset ominaisuudet eivät muuttuneet annettaessa samanaikaisesti lamivudiinia. Ei ole tietoa suurempien TMP/SMX-annosten, kuten PCP:n hoidossa käytettävien annosten, vaikutuksesta lamivudiinin farmakokinetiikkaan.
Zidovudiini
Kliinisesti merkittäviä muutoksia lamivudiinin tai tsidovudiinin farmakokinetiikassa ei havaittu 12 oireettomalla HIV-1-infektoituneella aikuisella, joille annettiin kerta-annos tsidovudiinia (200 mg) yhdessä useiden lamivudiiniannosten kanssa (300 mg 12 tunnin välein).
Mikrobiologia
Toimintamekanismi
Lamivudiini on synteettinen nukleosidianalogi. Solunsisäisesti lamivudiini fosforyloituu aktiiviseksi 5'-trifosfaattimetaboliitiksi, lamivudiinitrifosfaatiksi (3TC-TP). 3TC-TP:n pääasiallinen vaikutustapa on HIV-1:n käänteiskopioijaentsyymin (RT) esto DNA-ketjun lopettamisen kautta nukleotidianalogin liittämisen jälkeen.
Antiviraalinen toiminta
Lamivudiinin antiviraalinen aktiivisuus HIV-1:tä vastaan arvioitiin useissa solulinjoissa, mukaan lukien monosyytit ja tuoreet ihmisen perifeerisen veren lymfosyytit (PBMC) käyttäen tavanomaisia herkkyysmäärityksiä. EC50-arvot olivat välillä 0,003 - 15 mikroM (1 mikroM = 0,23 mcg per ml). Lamivudiinin EC50-arvojen mediaanit olivat 60 nM (alue: 20 - 70 nM), 35 nM (alue: 30 - 40 nM), 30 nM (alue: 20 - 90 nM), 20 nM (alue: 3 - 40 nM) , 30 nM (alue: 1 - 60 nM), 30 nM (alue: 20 - 70 nM), 30 nM (alue: 3 - 70 nM) ja 30 nM (alue: 20 - 90 nM) HIV-1-kladeja vastaan AG ja ryhmän O virukset (n = 3 paitsi n = 2 kladille B). EC50-arvot HIV-2-isolaatteja vastaan (n = 4) vaihtelivat välillä 0,003 - 0,120 mikroM PBMC:issä. Lamivudiini ei ollut antagonistinen kaikille testatuille HIV-lääkkeille. Kroonisen HCV-infektion hoidossa käytetty ribaviriini (50 mikroM) vähensi lamivudiinin anti-HIV-1-aktiivisuutta 3,5-kertaisesti MT-4-soluissa.
Resistanssi
HIV-1:n lamivudiiniresistenttejä variantteja on valittu soluviljelmässä. Genotyyppianalyysi osoitti, että resistenssi johtui spesifisestä aminohapposubstituutiosta HIV-1-käänteiskopioijaentsyymissä kodonissa 184, jolloin metioniini muuttui joko väliiniksi tai isoleusiiniksi (M184V/I).
HIV-1-kantoja, jotka ovat resistenttejä sekä lamivudiinille että tsidovudiinille, on eristetty koehenkilöistä. Kliinisten isolaattien herkkyyttä lamivudiinille ja tsidovudiinille seurattiin kontrolloiduissa kliinisissä tutkimuksissa. Potilailla, jotka saivat lamivudiinimonoterapiaa tai yhdistelmähoitoa lamivudiinin ja tsidovudiinin kanssa, useimpien koehenkilöiden HIV-1-isolaatit tulivat fenotyyppisesti ja genotyyppisesti resistenteiksi lamivudiinille 12 viikon kuluessa.
Hoidon aikana saadun HIV-1:n genotyyppi- ja fenotyyppianalyysi eristetään potilaista, joilla on virologinen epäonnistuminen
Kokeilu EPV20001
Viisikymmentäkolme 554:stä (10 %) EPV20001:een ilmoittautuneesta koehenkilöstä tunnistettiin virologisesti epäonnistuneiksi (plasman HIV-1 RNA -taso suurempi tai yhtä suuri kuin 400 kopiota/ml) viikkoon 48 mennessä. 28 potilasta satunnaistettiin saamaan lamivudiinia kerran. päivittäinen hoitoryhmä ja 25 lamivudiinia kahdesti päivässä saavaan hoitoryhmään. Keskimääräiset plasman HIV-1 RNA:n lähtötasot potilailla, jotka saivat lamivudiinia kerran vuorokaudessa ja lamivudiinia kahdesti vuorokaudessa saaneessa ryhmässä, olivat 4,9 log10 kopiota/ml ja 4,6 log10 kopiota/ml.
Hoidon aikana saatujen isolaattien genotyyppianalyysi 22 potilaalta, jotka tunnistettiin virologisesti epäonnistuneiksi lamivudiinia kerran päivässä saaneessa ryhmässä, osoitti, että isolaatit 8:sta 22:sta koehenkilöstä sisälsivät hoitoon liittyvän lamivudiiniresistenssiin liittyvän substituution (M184V tai M184I), isolaatit 0:sta 22:sta. koehenkilöt sisälsivät tsidovudiiniresistenssiin liittyviä hoidon aiheuttamia aminohapposubstituutioita (M41L, D67N, K70R, L210W, T215Y/F tai K219Q/E), ja isolaatit 10:stä 22:sta koehenkilöstä sisälsivät hoidon aiheuttamia aminohapposubstituutioita, jotka liittyivät efavirentsiresistenssiin ( L100I, K101E, K103N, V108I tai Y181C).
Hoidon aikana saatujen isolaattien genotyyppianalyysi koehenkilöistä (n = 22) kahdesti päivässä lamivudiinihoitoa saaneessa ryhmässä osoitti, että isolaatit 5:stä 22:sta koehenkilöstä sisälsivät hoidon aikana ilmeneviä lamivudiiniresistenssin substituutioita, isolaatit yhdestä 22:sta koehenkilöstä sisälsivät hoidon alkavaa tsidovudiiniresistenssiä. substituutiot, ja isolaatit 7:ltä 22:sta koehenkilöstä sisälsivät hoidon aiheuttamia efavirentsiresistenssisubstituutioita.
Lamivudiinia kerran vuorokaudessa saaneiden tutkimushenkilöiden (n = 13) lähtötilanteeseen sopivien HIV-1-isolaattien fenotyyppianalyysi osoitti, että isolaatit 7:stä 13:sta koehenkilöstä osoittivat 85-299-kertaisen alenemisen lamivudiiniherkkyydessä ja 12:sta. 13 koehenkilöä oli herkkiä tsidovudiinille, ja isolaateilla 8:sta 13:sta koehenkilön herkkyys efavirentsille pieneni 25-295-kertaisesti.
Lamivudiinia kahdesti vuorokaudessa saaneiden koehenkilöiden (n = 13) hoidon aikana saatujen lähtötasoon vastaavien HIV-1-isolaattien fenotyyppianalyysi osoitti, että 4:llä 13:sta koehenkilöstä saatujen isolaattien lamivudiiniherkkyys väheni 29-159-kertaisesti, isolaatit kaikista 13:sta. Tutkimushenkilöt olivat herkkiä tsidovudiinille, ja isolaatit 3:sta 13:sta koehenkilöstä osoittivat 21-342-kertaisen efavirentsin herkkyyden laskua.
Kokeilu EPV40001
Viisikymmentä henkilöä sai lamivudiinia 300 mg kerran vuorokaudessa plus tsidovudiinia 300 mg kahdesti vuorokaudessa sekä abakaviiria 300 mg kahdesti vuorokaudessa ja 50 koehenkilöä sai lamivudiinia 150 mg plus tsidovudiinia 300 mg ja abakaviiria 300 mg kaikki kahdesti vuorokaudessa. Keskimääräiset plasman HIV-1-RNA-tasot molemmissa ryhmissä olivat 4,79 log10 kopiota millilitraa kohti ja 4,83 log10 kopiota millilitraa kohti. Neljätoista 50 potilaasta lamivudiinia kerran päivässä saaneesta hoitoryhmästä ja 9 potilaasta 50 koehenkilöstä lamivudiinia kahdesti päivässä saaneessa ryhmässä todettiin virologisesti epäonnistuneiksi.
Hoidon aikana saatujen HIV-1-isolaattien genotyyppianalyysi koehenkilöistä (n = 9) kerran päivässä lamivudiinihoitoa saaneessa ryhmässä osoitti, että 6 koehenkilön isolaateissa oli pelkkä abakaviiri- ja/tai lamivudiiniresistenssiin liittyvä substituutio M184V. Hoidon aikana saadut isolaatit koehenkilöistä (n = 6), jotka saivat lamivudiinia kahdesti päivässä, osoittivat, että kahdelta koehenkilöltä saadut isolaatit saivat pelkän M184V:n, ja kahdelta koehenkilöltä saadut isolaatit sisälsivät M184V-substituution yhdessä tsidovudiiniresistenssiin liittyvien aminohapposubstituutioiden kanssa.
Lamivudiinia kerran vuorokaudessa saaneiden potilaiden (n = 6) hoidon aikana saatujen isolaattien fenotyyppianalyysi osoitti, että neljän henkilön HIV-1-isolaattien lamivudiiniherkkyys pieneni 32-53-kertaisesti. Näiden kuuden henkilön HIV-1-isolaatit olivat herkkiä tsidovudiinille.
Lamivudiinia kahdesti vuorokaudessa saavien potilaiden (n = 4) hoidon aikana saatujen isolaattien fenotyyppianalyysi osoitti, että yhdeltä koehenkilöltä peräisin olevien HIV-1-isolaattien lamivudiiniherkkyys pieneni 45-kertaisesti ja herkkyys tsidovudiinille pieneni 4,5-kertaisesti.
Pediatria
Lapsipotilaat, jotka saivat lamivudiinioraaliliuosta samanaikaisesti muiden antiretroviraalisten oraalisten liuosten (abakaviiri, nevirapiini/efavirentsi tai tsidovudiini) kanssa ARROW:ssa, kehittivät virusresistenssin useammin kuin niille, jotka saivat tabletteja. Kun EPIVIR ja abakaviiri annettiin kerran päivässä tai kahdesti vuorokaudessa, satunnaistettiin 13 %:lla potilaista, jotka aloittivat tablettien käytön, ja 32 %:lla potilaista, jotka aloittivat liuoksen käytön. Pediatriassa havaittu resistenssiprofiili on samanlainen kuin aikuisilla havaittujen genotyyppisten substituutioiden ja suhteellisen esiintymistiheyden suhteen, ja yleisimmin havaitut substituutiot ovat M184 (V tai I) [ks. Kliiniset tutkimukset ].
Ristivastus
Ristiresistenssiä on havaittu nukleosidikäänteiskopioijaentsyymin estäjillä (NRTI:t). Lamivudiiniresistentit HIV-1-mutantit olivat ristiresistenttejä didanosiinille (ddI) soluviljelmässä. Ristiresistenssi on odotettavissa myös abakaviirin ja emtrisitabiinin kanssa, koska ne valitsevat M184V-substituutiot.
Kliiniset tutkimukset
EPIVIRin käyttö perustuu kliinisten tutkimusten tuloksiin HIV-1-tartunnan saaneilla potilailla yhdistelmähoitona muiden antiretroviraalisten aineiden kanssa. Tiedot tutkimuksista, joissa on kliinisiä päätepisteitä tai CD4+-solumäärän ja HIV-1-RNA-mittausten yhdistelmää, on sisällytetty alla dokumentoimaan lamivudiinin vaikutusta yhdistelmähoitoon kontrolloiduissa tutkimuksissa.
Aikuiset aiheet
Kliininen päätepistetutkimus
NUCB3007 (CAESAR) oli monikeskus, kaksoissokkoutettu, lumekontrolloitu tutkimus, jossa verrattiin jatkuvaa nykyistä hoitoa (pelkästään tsidovudiinia [62 % koehenkilöistä] tai tsidovudiinia didanosiinin tai tsalsitabiinin kanssa [38 % koehenkilöistä]) EPIVIR 150 mg:n tai EPIVIR:n lisäämiseen. 150 mg plus tutkittava ei-nukleosidikäänteiskopioijaentsyymin estäjä (NNRTI), satunnaistettu 1:2:1. Mukaan otettiin yhteensä 1 816 HIV-1-infektoitunutta aikuista, joilla oli lähtötilanteessa 25–250 CD4+-solua/mm³ (mediaani = 122 solua/mm³): mediaani-ikä oli 36 vuotta, 87 % miehiä, 84 % nukleosideja kokeneita ja 16 % oli terapiattomia. Kokeen mediaanikesto oli 12 kuukautta. Tulokset on koottu taulukkoon 9.
Surrogate Endpoint Trials
Dual Nucleosid Analogue Trials
Tärkeimmissä kliinisissä tutkimuksissa lamivudiinin alkuvaiheessa verrattiin lamivudiini/tsidovudiini-yhdistelmiä tsidovudiinimonoterapiaan tai tsidovudiiniin ja tsalsitabiiniin. Nämä tutkimukset osoittivat lamivudiinin antiviraalisen vaikutuksen kahden lääkkeen yhdistelmänä. Lamivudiinin uudemmissa käytöissä HIV-1-infektion hoidossa se on sisällytetty useisiin lääkkeisiin, jotka sisältävät vähintään 3 antiretroviraalista lääkettä viruksen suppression tehostamiseksi.
Annostusohjelman vertailu Korvauspäätepistetutkimukset terapiattomia aikuisia
EPV20001 oli monikeskus, kaksoissokkoutettu, kontrolloitu tutkimus, jossa koehenkilöt satunnaistettiin 1:1 saamaan EPIVIR 300 mg kerran vuorokaudessa tai EPIVIR 150 mg kahdesti vuorokaudessa yhdessä tsidovudiinin 300 mg kahdesti vuorokaudessa ja efavirentsin 600 mg kerran vuorokaudessa kanssa. Mukaan otettiin yhteensä 554 antiretroviraalista hoitoa saamatonta HIV-1-tartunnan saanutta aikuista: mies (79 %), valkoinen (50 %), mediaani-ikä 35 vuotta, CD4+-solujen lähtöarvot 69-1089 solua/mm³ (mediaani = 362 solua per mm³) ja plasman HIV-1-RNA:n mediaani perustason 4,66 log10 kopiota ml:aa kohti. Kuvassa 1 ja taulukossa 10 on yhteenveto 48 viikon hoidon tuloksista.
Kuva 1: Virologinen vaste viikolle 48, EPV20001a,b (Intent-to-Treat) 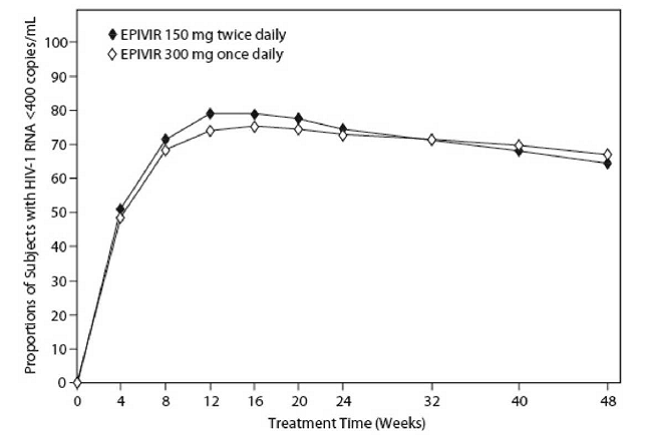
Roche AMPLICOR HIV-1 MONITORIN. b Vastaajia kullakin käynnillä ovat koehenkilöt, jotka olivat saavuttaneet ja säilyttäneet HIV-1 RNA:n alle 400 kopiota millilitraa kohden ilman, että hoito keskeytettiin kyseiseen käyntiin mennessä.
Niiden potilaiden osuudet, joilla HIV-1 RNA:ta oli alle 50 kopiota millilitrassa (Roche Ultrasensitive -määrityksellä) viikolle 48, oli 61 % potilaista, jotka saivat EPIVIR 300 mg kerran päivässä ja 63 % potilaista, jotka saivat EPIVIR 150 mg kahdesti vuorokaudessa. CD4+-solujen määrän mediaanilisäys oli 144 solua/mm³ viikolla 48 potilailla, jotka saivat EPIVIRiä 300 mg kerran vuorokaudessa, ja 146 solua/mm³ potilailla, jotka saivat EPIVIRiä 150 mg kahdesti vuorokaudessa.
Pieni, satunnaistettu, avoin pilottitutkimus, EPV40001, suoritettiin Thaimaassa. Mukaan otettiin yhteensä 159 aiemmin hoitamatonta aikuista henkilöä (mies 32 %, aasialainen 100 %, mediaani-ikä 30 vuotta, lähtötilanteen mediaani CD4+-solumäärä 380 solua/mm³, plasman HIV-1 RNA:n mediaani 4,8 log10 kopiota/ml). Tämän tutkimuksen kahdessa hoitohaarassa verrattiin lamivudiinia 300 mg kerran vuorokaudessa (n = 54) ja lamivudiinia 150 mg kahdesti vuorokaudessa (n = 52), kumpikin yhdistelmänä tsidovudiinin 300 mg kahdesti vuorokaudessa ja abakaviirin 300 mg kahdesti vuorokaudessa kanssa. 48 viikon tietojen intent-to-treat -analyyseissä potilaiden osuus, joilla HIV-1 RNA:ta oli alle 400 kopiota/ml, oli 61 % (33/54) ryhmässä, joka oli satunnaistettu saamaan kerran päivässä lamivudiinia, ja 75 % (39 52) ryhmässä, joka on satunnaistettu saamaan kaikki 3 lääkettä kahdesti päivässä; HIV-1 RNA:n osuudet alle 50 kopiota ml:ssa olivat 54 % (29/54) kerran päivässä saaneessa lamivudiiniryhmässä ja 67 % (35/52) kaikki kahdesti päivässä saaneessa ryhmässä; ja CD4+-solujen lukumäärän mediaanilisäys oli 166 solua/mm³ kerran vuorokaudessa saaneessa lamivudiiniryhmässä ja 216 solua/mm³ kaikki kahdesti päivässä saaneessa ryhmässä.
Pediatriset aiheet
Kliininen päätepistetutkimus
ACTG300 oli monikeskus, satunnaistettu kaksoissokkotutkimus, jossa vertailtiin EPIVIRiä ja RETROVIRia (tsidovudiinia) didanosiinimonoterapian kanssa. Näihin kahteen hoitoryhmään otettiin yhteensä 471 oireellista, HIV-1-infektoitunutta lapsipotilasta, jotka eivät olleet aiemmin saaneet hoitoa (vähemmän kuin 56 päivää antiretroviraalista hoitoa). Mediaani-ikä oli 2,7 vuotta (vaihteluväli: 6 viikosta 14 vuoteen), 58 % oli naisia ja 86 % ei-valkoisia. Keskimääräinen lähtötason CD4+-solumäärä oli 868 solua/mm³ (keskiarvo: 1060 solua/mm³ ja vaihteluväli: 0-4650 solua/mm³ alle 5-vuotiailla koehenkilöillä; keskiarvo: 419 solua/mm³ ja vaihteluväli: 0-1555 soluja/mm³ yli 5-vuotiaille koehenkilöille) ja keskimääräinen lähtötason plasma HIV-1 RNA oli 5,0 log10 kopiota/ml. Tutkimuksen keston mediaani oli 10,1 kuukautta potilailla, jotka saivat EPIVIR 150 mg plus RETROVIR, ja 9,2 kuukautta koehenkilöillä, jotka saivat didanosiinimonoterapiaa. Tulokset on koottu taulukkoon 11.
Kerran päivittäinen annostelu
ARROW (COL105677) oli viisi vuotta kestänyt satunnaistettu, monikeskustutkimus, jossa arvioitiin useita HIV-1-infektion kliinisen hoidon näkökohtia lapsilla. HIV-1-tartunnan saaneet, aiemmin hoitamattomat 3 kuukauden–17-vuotiaat tutkimushenkilöt otettiin mukaan ensimmäisen linjan hoito-ohjelmalla, joka sisälsi EPIVIRiä 150 mg ja abakaviiria kahdesti päivässä Maailman terveysjärjestön suositusten mukaisesti. Vähintään 36 viikon hoidon jälkeen koehenkilöille annettiin mahdollisuus osallistua ARROW-tutkimuksen satunnaistukseen 3, jossa verrattiin kerran vuorokaudessa annostelun turvallisuutta ja tehokkuutta EPIVIRin ja abakaviirin kahdesti vuorokaudessa annettuun annokseen yhdessä kolmannen antiretroviraalisen lääkkeen kanssa. lääkettä vielä 96 viikon ajan. 1 206 alkuperäisestä ARROW-potilaasta 669 osallistui satunnaistukseen 3. Virologinen suppressio ei ollut osallistumisen edellytys: satunnaistuksen 3 lähtötilanteessa (vähintään 36 viikon kahdesti päivässä suoritetun hoidon jälkeen) 75 % koehenkilöistä kahdesti päivässä. kohortti oli virologisesti tukahdutettu, kun vastaava luku oli 71 % kerran päivässä saaneesta kohortista.
Niiden potilaiden osuus, joiden HIV-1 RNA:ta oli alle 80 kopiota ml:ssa 96 viikon ajan, on esitetty taulukossa 12. Virologisten vasteiden väliset erot kahdessa hoitohaarassa olivat vertailukelpoisia sukupuolen ja iän perusteella.
Formulaatioanalyysit osoittivat niiden potilaiden osuuden, joiden HIV-1 RNA:ta oli alle 80 kopiota/ml satunnaistuksen yhteydessä, ja viikko 96 oli suurempi potilailla, jotka olivat saaneet EPIVIR 150 mg:n ja abakaviirin tablettiformulaatioita (75 % [458/610] ja 72 % [434/601]) kuin niillä, jotka olivat saaneet liuosvalmisteita (EPIVIR 150 mg liuosta annettuna painoaluekohtaisina annoksina noin 8 mg/kg/vrk) milloin tahansa (52 % [29/56] ja 54 % [30/56]), vastaavasti [katso VAROITUKSET JA VAROTOIMET ]. Nämä erot havaittiin kussakin eri arvioidussa ikäryhmässä.
POTILASTIEDOT
EPIVIR (EP-i-veer) (lamivudiini) tabletit
EPIVIR (EP-i-veer) (lamivudiini) oraaliliuos
Mikä on tärkein tieto, joka minun pitäisi tietää EPIVIR 150mg:sta?
EPIVIR voi aiheuttaa vakavia sivuvaikutuksia, mukaan lukien:
- Hepatiitti B -viruksen paheneminen ihmisillä, joilla on HIV-1-infektio. Jos sinulla on HIV-1 (ihmisen immuunikatovirus tyyppi 1) ja hepatiitti B -virus (HBV), HBV-virus voi pahentua (viruksen puhkeaminen), jos lopetat EPIVIRin käytön. "Leimaus" on, kun HBV-infektiosi yhtäkkiä palaa huonommalla tavalla kuin ennen. Maksasairauden paheneminen voi olla vakavaa ja johtaa kuolemaan.
- EPIVIR ei lopu kesken. Täytä reseptisi tai keskustele terveydenhuollon tarjoajasi kanssa ennen kuin EPIVIR on loppunut.
- Älä lopeta EPIVIR-hoitoa keskustelematta ensin terveydenhuollon tarjoajan kanssa.
- Jos lopetat EPIVIRin käytön, terveydenhuollon tarjoajasi tulee tarkistaa terveytesi usein ja tehdä verikokeita säännöllisesti useiden kuukausien ajan maksan tarkistamiseksi.
- Resistentti hepatiitti B -virus (HBV). Jos sinulla on HIV-1 ja hepatiitti B, hepatiitti B -virus voi muuttua (mutatoitua) EPIVIR 150 mg -hoidon aikana ja muuttua vaikeammaksi hoidettavaksi (resistentiksi).
- Käytä interferoni- ja ribaviriinipohjaisten hoito-ohjelmien kanssa. Kuoleman aiheuttaneen maksasairauden pahenemista on tapahtunut sekä HIV-1- että hepatiitti C -tartunnan saaneilla henkilöillä, jotka käyttävät antiretroviraalisia lääkkeitä ja joita myös hoidetaan hepatiitti C:n vuoksi interferonilla ribaviriinin kanssa tai ilman. Jos käytät EPIVIR 150 mg ja interferonia ribaviriinin kanssa tai ilman, kerro terveydenhuollon tarjoajallesi, jos sinulla on uusia oireita.
Mikä on EPIVIR?
EPIVIR on reseptilääke, jota käytetään yhdessä muiden antiretroviraalisten lääkkeiden kanssa ihmisen immuunikatovirus (HIV-1) -infektion hoitoon.
HIV-1 on virus, joka aiheuttaa hankitun immuunikato-oireyhtymän (AIDS).
EPIVIR-tabletit ja oraaliliuos (käytetään HIV-1-infektion hoitoon) sisältävät suuremman annoksen samaa vaikuttavaa ainetta (lamivudiinia) kuin lääkkeessä EPIVIR-HBV-tabletit ja oraaliliuos (käytetään HBV:n hoitoon). Jos sinulla on sekä HIV-1 että HBV, sinun ei tule käyttää EPIVIR-HBV:tä infektioidesi hoitoon.
EPIVIRin turvallisuutta ja tehoa ei ole osoitettu alle 3 kuukauden ikäisillä lapsilla.
Kuka ei saa ottaa EPIVIR 150 mg -valmistetta?
Älä ota EPIVIRiä jos olet allerginen lamivudiinille tai jollekin EPIVIRin aineosalle. Katso tämän potilastiedotteen lopusta täydellinen luettelo EPIVIRin ainesosista.
Mitä minun pitäisi kertoa terveydenhuollon tarjoajalleni ennen EPIVIRin ottamista?
Ennen kuin otat EPIVIR 150 mg -valmistetta, kerro terveydenhuollon tarjoajallesi, jos:
- sinulla on tai on ollut maksaongelmia, mukaan lukien B- tai C-hepatiittivirusinfektio.
- on munuaisongelmia.
- on diabetes. Jokainen 15 ml:n annos (150 mg) EPIVIR-oraaliliuosta sisältää 3 grammaa sakkaroosia.
- olet raskaana tai suunnittelet raskautta. EPIVIRin käyttöön raskauden aikana ei ole yhdistetty lisääntynyttä synnynnäisten epämuodostumien riskiä. Keskustele terveydenhuollon tarjoajan kanssa, jos olet raskaana tai suunnittelet raskautta. Raskausrekisteri. On olemassa raskausrekisteri naisille, jotka käyttävät antiretroviraalisia lääkkeitä raskauden aikana. Tämän rekisterin tarkoituksena on kerätä tietoja sinun ja vauvasi terveydestä. Keskustele terveydenhuollon tarjoajasi kanssa siitä, kuinka voit osallistua tähän rekisteriin.
- imetät tai aiot imettää. Älä imetä, jos käytät EPIVIRiä.
- Älä imetä, jos sinulla on HIV-1, koska sinulla on riski siirtää HIV-1 lapsellesi.
Kerro terveydenhuollon tarjoajallesi kaikista käyttämistäsi lääkkeistä, mukaan lukien resepti- ja käsikauppalääkkeet, vitamiinit ja yrttilisät.
Jotkut lääkkeet ovat vuorovaikutuksessa EPIVIRin kanssa. Pidä luetteloa lääkkeistäsi ja näytä se terveydenhuollon tarjoajalle ja apteekkihenkilökunnalle, kun hankit uuden lääkkeen. Voit pyytää terveydenhuollon tarjoajaltasi tai apteekista luettelon lääkkeistä, joilla on vuorovaikutus EPIVIRin kanssa.
Älä aloita uuden lääkkeen käyttöä kertomatta asiasta terveydenhuollon tarjoajallesi. Terveydenhuollon tarjoaja voi kertoa sinulle, onko EPIVIRin ottaminen muiden lääkkeiden kanssa turvallista.
Miten minun pitäisi ottaa EPIVIR?
- Ota EPIVIR juuri sen verran kuin terveydenhuollon tarjoajasi on neuvonut sinua ottamaan sen.
- Jos unohdat ottaa EPIVIR 150 mg -annoksen, ota se heti kun muistat. Älä ota kahta annosta samanaikaisesti tai ota enempää kuin mitä terveydenhuollon tarjoajasi on määrännyt.
- Pysy terveydenhuollon tarjoajan hoidossa EPIVIR-hoidon aikana.
- EPIVIR voidaan ottaa ruoan kanssa tai ilman.
- Kolmen kuukauden ikäisille ja sitä vanhemmille lapsille terveydenhuollon tarjoaja määrää EPIVIR 150 mg -annoksen lapsesi painon perusteella.
- Kerro terveydenhuollon tarjoajallesi, jos sinulla tai lapsellasi on vaikeuksia niellä tabletteja. EPIVIR 150mg on myös nestemäinen (oraaliliuos).
- Älä lopu EPIVIRistä. Viruksen määrä veressäsi voi lisääntyä ja virusta voi olla vaikeampi hoitaa. Kun tarjontasi alkaa olla vähissä, hanki lisää terveydenhuollon tarjoajalta tai apteekista.
- Jos otat liikaa EPIVIR 150 mg -valmistetta, soita välittömästi terveydenhuollon tarjoajalle tai mene lähimmän sairaalan ensiapuun.
Mitkä ovat EPIVIR 150mg:n mahdolliset sivuvaikutukset?
- EPIVIR 150mg voi aiheuttaa vakavia sivuvaikutuksia, mukaan lukien:
- Katso "Mikä on tärkein tieto, joka minun pitäisi tietää EPIVIR 150mg:sta?"
- Hapon kertyminen vereen (maitohappoasidoosi). Maitohappoasidoosia voi esiintyä joillakin EPIVIR-hoitoa käyttävillä ihmisillä. Maitohappoasidoosi on vakava lääketieteellinen hätätilanne, joka voi aiheuttaa kuoleman. Soita välittömästi terveydenhuollon tarjoajallesi, jos saat jonkin seuraavista oireista, jotka voivat olla maitohappoasidoosin merkkejä:
- tuntuu erittäin heikolta tai väsyneeltä
- tuntuu kylmältä, etenkin käsissäsi ja jaloissasi
- epätavallinen (ei normaali) lihaskipu
- huimausta tai pyörrytystä
- vaikeuksia hengittää
- sinulla on nopea tai epäsäännöllinen syke
- vatsakipu, johon liittyy pahoinvointia ja oksentelua
- Vakavia maksaongelmia voi tapahtua ihmisillä, jotka käyttävät EPIVIRiä. Joissakin tapauksissa nämä vakavat maksaongelmat voivat johtaa kuolemaan. Maksasi voi kasvaa suureksi (hepatomegalia) ja maksassasi voi kehittyä rasvaa (steatoosi). Soita välittömästi terveydenhuollon tarjoajallesi, jos saat jonkin seuraavista maksaongelmien merkeistä tai oireista:
- ihosi tai silmiesi valkoinen osa muuttuu keltaisiksi (keltatauti)
- ruokahaluttomuus useita päiviä tai pidempään
- pahoinvointi
- tumma tai "teenvärinen" virtsa
- kipua, särkyä tai arkuutta vatsan oikealla puolella
- vaaleat ulosteet (suolen liikkeet)
Saatat saada todennäköisemmin maitohappoasidoosin tai vakavia maksaongelmia, jos olet nainen tai olet erittäin ylipainoinen (lihava).
- Haimatulehduksen (haimatulehduksen) riski. Lapsilla saattaa olla riski saada haimatulehdus EPIVIR-hoidon aikana, jos he:
- olet käyttänyt nukleosidianalogisia lääkkeitä
- sinulla on aiemmin ollut haimatulehdus
- sinulla on muita haimatulehduksen riskitekijöitä
Soita välittömästi terveydenhuollon tarjoajallesi, jos lapsellesi kehittyy haimatulehduksen merkkejä ja oireita, mukaan lukien vaikea ylävatsan alueen kipu, pahoinvoinnin ja oksentelun kanssa tai ilman. Terveydenhuollon tarjoaja voi kehottaa sinua lopettamaan EPIVIR 150 mg:n antamisen lapsellesi, jos hänen oireensa ja verikoetulokset osoittavat, että lapsellasi saattaa olla haimatulehdus.
- Muutokset immuunijärjestelmässäsi (immuunireaktion oireyhtymä) voi tapahtua, kun aloitat HIV-1-lääkkeiden käytön. Immuunijärjestelmäsi voi vahvistua ja alkaa taistella infektioita vastaan, jotka ovat olleet piilossa kehossasi pitkään. Kerro heti terveydenhuollon tarjoajallesi, jos sinulla alkaa ilmaantua uusia oireita EPIVIR-hoidon aloittamisen jälkeen.
EPIVIR 150mg:n yleisimmät sivuvaikutukset aikuisilla ovat:
- päänsärky
- nenän merkit ja oireet
- pahoinvointi
- ripuli
- ei yleensä voi hyvin
- väsymys
- yskä
EPIVIR 150mg:n yleisimmät sivuvaikutukset lapsilla ovat kuume ja yskä.
Kerro terveydenhuollon tarjoajallesi, jos sinulla on jokin haittavaikutus, joka häiritsee sinua tai joka ei katoa.
Nämä eivät ole kaikkia EPIVIRin mahdollisia sivuvaikutuksia. Soita lääkärillesi saadaksesi lääketieteellisiä neuvoja sivuvaikutuksista. Voit ilmoittaa sivuvaikutuksista FDA:lle numerossa 1-800-FDA-1088.
Kuinka EPIVIR tulee säilyttää?
- Säilytä EPIVIR 150 mg tabletteja ja oraaliliuosta huoneenlämmössä 20 °C - 25 °C (68–77 °F).
- Pidä EPIVIR 150 mg oraaliliuoksen pullot tiiviisti suljettuina.
Pidä EPIVIR ja kaikki lääkkeet poissa lasten ulottuvilta.
Yleistä tietoa EPIVIRin turvallisesta ja tehokkaasta käytöstä.
Lääkkeitä määrätään joskus muihin tarkoituksiin kuin potilasohjeessa lueteltuihin tarkoituksiin. Älä käytä EPIVIRiä sairauteen, johon sitä ei ole määrätty. Älä anna EPIVIRiä muille ihmisille, vaikka heillä olisi samat oireet kuin sinulla. Se voi vahingoittaa heitä.
Voit kysyä terveydenhuollon tarjoajaltasi tai apteekista tietoja EPIVIR 150mg -valmisteesta, joka on kirjoitettu terveydenhuollon ammattilaisille.
Lisätietoja saat osoitteesta www.viivhealthcare.com tai soittamalla numeroon 1-877-844-8872.
Mitä ainesosia EPIVIR 150mg sisältää?
Vaikuttava aine: lamivudiini
Inaktiiviset ainesosat:
EPIVIR 150 mg jakouurteiset 150 mg kalvopäällysteiset tabletit: hypromelloosi, magnesiumstearaatti, mikrokiteinen selluloosa, polyetyleeniglykoli, polysorbaatti 80, natriumtärkkelysglykolaatti ja titaanidioksidi.
EPIVIR 300 mg kalvopäällysteiset tabletit: musta rautaoksidi, hypromelloosi, magnesiumstearaatti, mikrokiteinen selluloosa, polyetyleeniglykoli, polysorbaatti 80, natriumtärkkelysglykolaatti ja titaanidioksidi.
EPIVIR 150 mg oraaliliuos: keinotekoiset mansikka- ja banaaniaromit, sitruunahappo (vedetön), metyyliparabeeni, propyleeniglykoli, propyyliparabeeni, natriumsitraatti (dihydraatti) ja sakkaroosi (200 mg/ml).
Yhdysvaltain elintarvike- ja lääkevirasto on hyväksynyt nämä potilastiedot.